Evaluation of Microscopic Analytical Techniques for the Analysis of Artists’ Materials
Abstract
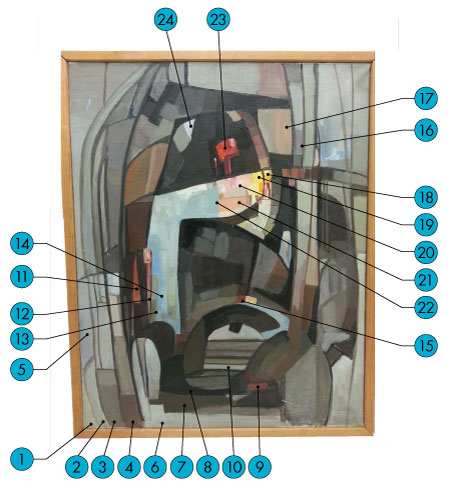
My senior thesis utilized samples from a painting to determine which instrumental techniques are most appropriate and effective for the identification of artists’ materials. I analyzed a 30” X 23.75” abstract study painting given to me by the Hooke College of Applied Science’s pigment instructor, Joe Barabe. In my study I took 24 samples of different colors from the painting (Figure 1). From there used each of the samples in different forms of analysis including polarized light microscopy (PLM), scanning electron microscopy with energy dispersive x-ray spectrometry (SEM/EDS), Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy and x-ray diffraction (XRD). Once I collected the data from each of these forms of analysis, I compared the results to identify each of the pigments taken from the painting, and determined which method or combination of methods was most effective for each material. Advantages and disadvantages of each method in relation to artists’ pigments were also discussed.
Background
Works of art have been around since the beginning of time, from cave paintings to sculptures to portraits. It has been understood as a way of conveying something whether it is a story, a person, or an emotion. Despite the fact that art has been around for so long, the appreciation for the importance of these works of art has not always been held in as high regard as it is today. Now people appreciate it for not only its aesthetic value, but its historical insight as well.
There is much dispute as to when art restoration truly began in Europe, but one date that is agreed upon by most is 1565 when the Sistine Chapel was first restored (Caple, 101). From this point on, until the early twentieth century, more often than not, artists were called upon to repair damaged artwork because they were more concerned with preserving the aesthetics of the piece rather than its originality. However, artists were not the only ones taking an interest in restoration during this time period. Scientists were also interested in understanding how and why pieces of art were deteriorating. One such scientist was Michael Faraday, who was hired by the National Gallery in London to investigate different cleaning methods for artwork. He conducted analytical and deterioration studies of art and found how sulfur compounds liberated from coal smoke and gas lighting damaged painting as well as how the increased humidity during London fogs increased deterioration (Stoner, 41). Another scientist who was also working at this time was Louis Pasteur, and he carried out analytical studies of paint and optical crystallography in the 1870s (Stoner, 41).
The late 1800s to the early 1900s was when the definition of art conservation began to change. Artists and art historians began to realize that science could be applied to preventing further deterioration and that it could be used for authentication as well as conserving the originality of a work of art. PLM was one of the first methods used for pigment analysis because it allowed for micro-sampling, which was as non-destructive as possible at the time. PLM was used to characterize different pigments, and then this information was used to identify pigments and give tentative dates of creation based on pigments used. As the years progressed, so did scientific techniques used in art conservation. Some of the main instruments used today are FTIR, SEM-EDS, Raman spectroscopy and XRD. An example of using multiple techniques for analysis is the “Ormylia” Foundation, Art Diagnosis Centre in Greece where they used all of these techniques and more to examine Byzantine Icons from the fifteenth to eighteenth centuries. They wanted to identify the pigments in the different layers of paint and distinguish between what was from the original painting of the icon and what has been added since. They also wanted to identify the different binders used in the paints. The “Ormylia” Foundation used micro-Raman spectroscopy and SEM-EDS to identify the pigments in a carefully cut cross section. Raman is very sensitive to inorganic compounds, which is why it is so useful for older paintings since they were made from minerals, however, SEM-EDS helps confirm the Raman findings because it is used to identify morphology and elements present. Additionally, micro-FTIR was also used to find the different binding media in the paint layers because those are generally organic compounds, which FTIR is most adept at determining. Understanding what binding media is used is important in comprehending the Byzantine artists’ painting techniques (Sotiropoulou and Sister Danilia, 877). Overall, this study is a prime example of how different techniques can be used in art conservation, as well as how they can be used together to support one another to discover something that was not previously known. Similar to the above study, I used PLM, XRD, SEM equipped with EDS detector, FTIR and Raman spectroscopy to identify pigments in an abstract painting. While performing the different forms of analysis I also noted how each technique has its advantages and disadvantages, but putting together all the data from the different analyses allowed for a clear overall picture of the pigments used in the painting.
Bulk Sample Preparation
In order to obtain the bulk 24 samples from the painting, I used a stereomicroscope on a boom stand to reach the pigments towards the center of the painting. A boom stand allows the stereomicroscope to swivel, which permits easier access to all areas of the painting. I used a coarse tungsten needle to scrape microscopic amounts of paint off the painting and onto a prepared a slide. (Figure 2.) The slide holds my bulk sample, so I would have enough for more than one type of analysis. In order to prepare this slide I used a glass microscope slide, coverglass, and tape. I used the tape to attach the cover slip to the slide, which creates a protective section for the pigments to be stored.
Section 1: Polarized Light Microscopy
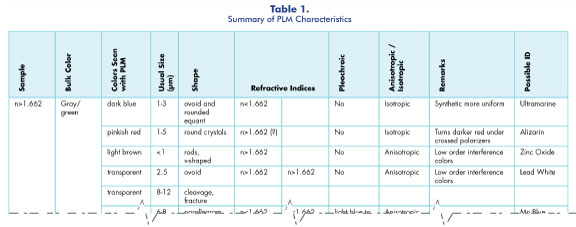
After each of the 24 samples was extracted from the painting with the least amount of destruction to the painting as possible, i.e. there were no macroscopic changes to the painting, each sample was mounted to a microscope slide using permanent mounting material. The permanent mounting medium that is available at Hooke College of Applied Sciences is called Meltmount®, which has a refractive index of 1.662. This is important to note because many of the references for identifying pigments compare the pigment’s refractive index relative to 1.662. A small glass rod is used to pick up a droplet of heated Meltmount and it is dropped onto a warmed glass slide. This was accomplished by heating the Meltmount on a laboratory hotplate until the heated mount dropped smoothly off the glass rod. Then a coarse tungsten needle is used to remove a tiny amount of particle from the bulk sample and place it onto the droplet of Meltmount on the glass slide. The Meltmount dries quickly, so once the sample is placed on the permanent mount, the glass slide is placed on the hotplate for about 10 seconds to re-melt the mount. Then the slide is quickly removed and a glass coverglass is pressed down on the sample, which crushes the pigments. This helps in spreading out the pigments so they are not all conglomerates and it allows the individual pigments to be seen more easily. Once all the samples were mounted on the slides, they were examined using an Olympus BX51 polarized light microscope. Additionally, photomicrographs were taken of each sample and can be seen in Appendix A.
Polarized light microscopy uses a light microscope equipped with transmitted, oblique and fluorescence illumination to characterize different types of material including pigments. Two polarizing agents are also on the microscope and these are used to observe characteristics that cannot be seen by ordinary light. Ordinary light is vibrating in all different directions, but when a polarizing filter is used on ordinary light it only allows light that is vibrating in one direction to pass through the filter (Van Howe, 42). The microscope is also equipped with a red-1-compensator, which when inserted will add or subtract 530 nm wavelengths of light which is, in turn, used to help confirm interference colors seen between crossed polarizers. Using PLM, one can observe a material’s refractive index, extinction, morphology, and size. Additionally, one can also determine whether a material is isotropic or anisotropic, as well as its crystallinity (Van Howe, 9-10).
Pigment 1: Light Brown/White
After examining each of the samples using PLM, I observed that there seemed to be 11 similar pigment types, mixed in different ratios, in order to produce the different colors. This observation will be confirmed using several other forms of analysis. The dominant pigment that was found in every sample taken from the painting was a light brown color in transmitted light. On closer inspection, however, the individual pigments were colorless with a slight brown tint on the outer surface of the particle. The pigment appeared a lighter shade of brown when aggregated. The pigment particles were individually very small—all less than 1 µm in diameter. This makes it extremely difficult to determine their morphology, other than that they are slightly roundish. Next, I used the crossed polarizers in order to determine if the brown pigment was anisotropic or isotropic. When I inserted the crossed polarizers the light brown pigment became bright, but I was unable to determine the extinction because the particles were so small. After that, I inserted my red-1-compensator, which allowed me to observe that the pigment exhibited first-order colors of the Michel-Lévy chart, specifically low-order grays and whites. A Michel-Lévy chart is used to determine the birefringence of a material. The chart uses the particle’s thickness and interference colors or colors seen using crossed polarizers to determine the birefringence. Next, I switched back to plane polarized light in order to determine the refractive index of the pigment; however, because of the size of the particles/pigments it was difficult to observe whether or not the Becke line was moving into or out of the particle when the stage was lowered using a 40X objective. Therefore, I used an oil immersion lens to get better resolution. I found that the Becke line moved into the pigment when the stage was lowered and I therefore concluded that the refractive index was greater than the mounting medium of 1.662. Once all of these characteristics were established, I used the flowchart for white pigments because they are often seen as the light brown color in transmitted light, given in the Pigment Identification Manual (Appendix A), to identify it. From the characteristics described above and from comparing different samples of known identity, I concluded that the white pigment found throughout the painting is zinc oxide.
Pigment 2: Transparent Ovoids
The second suspected white pigment that I found throughout the painting was most likely lead white. The pigment was about 2.5 μm in diameter and ovoid in shape with a dark outline in transmitted light. Next, I crossed the polarizers and found that the particle was anisotropic with low-order gray interference colors. I rotated the stage to an extinction position and switched back to transmitted light in order to examine the Becke line. I used my aperture diaphragm to increase the contrast between my particle and the Meltmount. I adjusted the fine focus so the stage moved down and I saw that the Becke line moved into the particle so the refractive index at that position was greater than 1.662. After that I rotated the stage 90 degrees and looked at the Becke line again at this extinction position and found that it too had a refractive index higher than 1.662. Using the flow chart found in the Pigment Identification Manual, the pigment that best matches these characteristics is lead white (Appendix A).
Pigment 3: Rose Red
The next pigment that I found in the majority of the samples was a rose red color in transmitted light. This is probably because it appeared to be mixed with yellow and white to create a variety of hues of brown that can be seen in Figure 1. Furthermore, the pigment varied in size from between 1 and 15 µm in diameter. This pigment also appeared more rounded than angular in appearance and also tended to have a slightly darker dot at the center of the pigment. When I crossed the polarizers the red appeared darker but it did not change at all in color when the stage was rotated. This could be because it was mixed with white and other pigments so it may be isotropic on its own. Next, I used the oil immersion lens on all the samples that contained the red pigments in order to determine the Becke line. I saw that the Becke lines moved into the particle when the stage was lowered and therefore the refractive index of the particle is higher than the Meltmount (1.662). Based on the color, morphology, isotropicism, and refractive index, I believe that this pigment was made from the madder plant. I think it most likely a variation of the alizarin lake pigment due to its rosy color and size. Additionally, according to Painting Materials: A Short Encyclopaedia, the deepness of the red can vary depending on what base is used to create the pigment.
Pigment 4: Dark Red
When I was first examining the different samples it appeared that there was only one red pigment throughout the painting. However, after I took a second and third look, I noticed that some of the red particles were darker and smaller than the rose red. Additionally, after I first mounted the samples, some of the pigments were not well dispersed in the medium; so, I reheated the sample on the hot plate and used a cuticle pusher to press down on the coverglass in order to get a better dispersion of the pigments. This allowed me to distinguish the second red pigment. This red pigment, when dispersed well, is about 1 μm in diameter; however it appeared larger when it was clumped together. The pigment’s shape was very small, rounded equants. When I crossed the polarizers the pigments appeared dark, therefore they were isotropic. I switched back to transmitted light and examined the Becke line to determine the relative refractive index. It was difficult to determine because the pigments were so small, but the line appeared to move in when the stage was lowered so the refractive index was higher than 1.662. Upon following the flowchart in the Pigment Identification Manual the pigment that best fit these characteristics was cadmium red (Appendix A).
Pigment 5: Golden Orange/Yellow
This pigment was found in brown bulk samples. It is a golden orange color in transmitted light and it was found mostly imbedded with the white pigment, which made it difficult to determine the size range. The size range that I found was between 5 and 20 µm. The structure of the pigment was rounded and slightly globular in appearance. When the polarizers were crossed it appeared a deeper orange with some anisotropic inclusions present that demonstrated undulose extinction. Next, the Becke line test was observed and the lines moved in when the stage was lowered so the refractive index is greater than 1.662. When combining these characteristics and using the Pigment Identification Manual and Painting Materials: A Short Encyclopaedia, the pigment that best matched these characteristics was yellow ochre (Appendix A).
Pigment 6: Warm Light Yellow
This pigment was found in the samples taken from rich brown, yellow, and orange sections of the painting. The pigment is very small and ranges in size from about 0.5 to 2.5 µm; however some may have been a bit larger, it was difficult to determine the exact size due to the fact that this pigment formed aggregates more often than not. It was a pale yellow in transmitted light, with a slightly darker outline and irregular shapes. When the Becke line test was implemented it was determined that the pigment had a refractive index greater than the medium at 1.662. After that was determined, I crossed the polarizers and the pigment appeared to be isotropic with anisotropic inclusions that demonstrated undulose extinction. Additionally, the pigment appeared a darker yellow—and in some places slightly green—under crossed polarizers. After consulting not only Painting Materials: A Short Encyclopaedia, but also the McCrone Atlas of Microscopic Particles, I believe that the characteristics of this pigment are consistent with cadmium yellow.
Pigment 7: Dark Yellow
Sample 18 from the bright yellow spot on the painting appeared to be a completely different yellow than the previous samples. The size range was very tiny—between 0.5 and 1 μm. The shape of this pigment was rounded equant; so, not quite circular but rounded edges and fairly even. When the polarizers were crossed the pigment appeared green, with some brighter inclusions. Lastly, the Becke line test was performed and it appeared that the line moved out as the stage was lowered indicating that the refractive index of the sample was less than 1.662. However, because the pigments were so tiny, it was very difficult to determine which way the Becke line went. Therefore, at this time this pigment could not be identified using solely PLM.
Pigment 8: Dark Blue
This next pigment was found in bulk samples that were gray or blue in color. It ranged in size from about 2 to 7 µm and its shape was generally flat equant with rounded edges. The blue was a dark medium blue that looked very clear with transmitted light. It was isotropic, so it did not appear using crossed polarizers. It did appear, however, with crossed polarizers and the red-1-compensator as a darker blue. In addition, the Becke line test was observed and it appeared that the particle had a refractive index of less than 1.662. After consulting the Painting Materials: A Short Encyclopaedia and the Atlas, the characteristics of the pigment appear to be consistent with ultramarine blue.
Pigment 9: Light Pleochroic Blue
This pigment was found in sample 14, which was taken from the sky blue bulk sample. It is a very pale light blue in transmitted light. It is also pleochroic and becomes almost transparent with a hint of blue when the stage is rotated. Additionally, it was anisotropic with parallel extinction. The pigment displayed first-order white/gray colors under crossed polarizers. This was confirmed when the red-1-compensator was inserted and it changed to blue and yellow when the stage was rotated. Next, the Becke line test was observed and it showed that the Becke lines moved in when the stage was lowered and therefore the refractive index is higher than that of the medium (1.662). After thorough examination of the characteristics described above, including morphology, the best blue pigment that matches is manganese blue.
Pigment 10: Light Green
This pigment was found in soft gray/green areas on the painting. It was found imbedded with the light brown pigment found throughout the painting. It looks sea foam green in transmitted light; however, it is also pleochroic so it goes from sea foam green to a more yellowish-green when the stage is rotated. I could not get the exact size range because it was so imbedded in the white pigment and it formed aggregates. The pigment does not appear coarse, but rather more crystalline and spherical in shape and appearance. It is anisotropic with second-order colors and higher. This was confirmed with the red-1-compensator. Furthermore, the pigment displayed undulose extinction under crossed polarizers. The becke line test was performed and it showed that the refractive index of the pigment was greater than that of the medium (1.662). At this point in my analysis I am not sure what the identity of this pigment is; however, I thought that it may be verdigris, but the refractive indices do not match what I saw in this pigment; although I wonder if the results were affected because it was so embedded in the whitening pigment. This could affect the results of the Becke line test because the Becke line moves to the medium with the highest refractive index when the stage is lowered; so if the green pigment was so embedded into the white, the Becke line might be responding to the white pigment refractive index and not the light green one.
Pigment 11: Opaque Black
This pigment was found in the very dark brown or black sections in most areas on the painting; however, there were some sections that were slightly lighter and appeared more gray than black. It appears to be mostly opaque, especially when it forms aggregates. When it was more dispersed it was slightly more transparent. It is a dark brown/black in transmitted light and, due to its opacity, it is isotropic under crossed polarizers. I was unable to determine a size range for the particles because they clumped so closely together; so, I could not clearly see the individual particles. The Becke line test was also conducted and it showed that the pigment’s refractive index was less than 1.662. At this time and based on the above characteristics, the pigment that best fits these characteristics is bone black.
{kind=link}
Section 2: X-ray Diffraction (XRD)
The next instrument that I used in my analysis was an x-ray diffraction system. In this instrument, x-rays are emitted from a beam to a sample, where, depending on the crystal structure of the sample, the x-rays are diffracted in specific patterns. These patterns can determine the crystal structure, which, in turn, can identify different compositions. One advantage of XRD is that it is a nondestructive form of analysis and therefore the samples used in this analysis can be used in other instruments as well. The specific instrument on which I ran my samples was a Rigaku Rapid-II. The first step that I took in this section of the analysis was mounting each of the 24 samples onto 24 MiTegen pins (MiTegen, Ithaca, NY). This was an extremely meticulous process; however, a large sample is not needed, which is an advantage since it is important to be as noninvasive as possible with respect to the painting. The sample size ranges from 20 to 100 µm. Therefore, I used the bulk samples prepared when originally sampling the painting for all my XRD mounts. The first step in the mounting procedure is to prepare all the instruments needed to mount the sample. The first thing that needs to be made is a soluble gum slide. To do this place a drop of amyl acetate onto a glass slide. Then take a piece of double-sided tape and place it top side down onto the amyl acetate solution on the glass slide. Let it sit for about 30 seconds and remove the piece of tape, what should remain on the slide is the adhesive from the tape. After that, take another glass slide and place a different piece of double-sided tape on it. Then place the MiTegen pin on the double-sided tape, this ensures that the pin will not roll when the sample is being put on it (Image 3).


Once these preparatory steps are completed then the actual mounting can commence. There are several different methods that can be used to mount the particle onto the MiTegen pin. The one that worked best for me began by using a stereomicroscope and a medium tungsten needle to make a microscopic ball of soluble gum and transport it to the very tip of the MiTegen pin. Then, using the same medium tungsten needle, I removed a small particle of paint from the bulk sample. I chose the particle that had the most colored pigments embedded it, so as to not only analyze the white pigment used as a ground layer throughout the painting. I carefully placed this particle on top of the soluble gum on the very tip of the pin (Image 2). The sample must be on the tip of the pin, so the pin can be aligned with the beam easier. Once the sample was mounted onto the pin correctly, I transported the pin to the XRD holder and inserted the pin into it. Then, I took the whole apparatus and placed it on the goniometer. Before I could run the sample, I had to center the pin and holder so, when it was rotated, the beam of x-rays always hit the sample. After this was accomplished, I ran each of the samples for 15 minutes.
Once the data was collected using the RINT Rapid software, I transported the file to the 2DP Theta Conversion software where I converted the data from an image of different intensity rings to two-theta/intensity spectrum, which is the standard format used in different libraries. I then transferred this new file to the Jade 9 software where I calibrated the spectrum to a silicon standard and ran a library search to identify the spectrum. The Jade software is linked the International Centre of Diffraction Database (ICDD). This database contains over 300,000 XRD reference patterns for comparison. More often than not, the library search would identify some of the peaks, but not all of them; so, I had to run a secondary search for the minor peaks to identify the second component of the spectrum. However, even with the secondary search, there are some cases that there were still unidentified peaks. Therefore, I decided to run my samples in the SEM-EDS instrument as well to narrow down the elemental composition. This helped narrow down the library search results since I could select certain elements that were present in the sample. Additionally, it is better to run my samples in the XRD first since I can remove them from the pin mount once the analysis is done, and then mount them for the SEM without damaging the sample. Whereas, if the particle was put in the SEM first it could have some beam damage from the electron beam. The table below shows a summary of all my results observed for each of my samples after the SEM-EDS data was consulted.
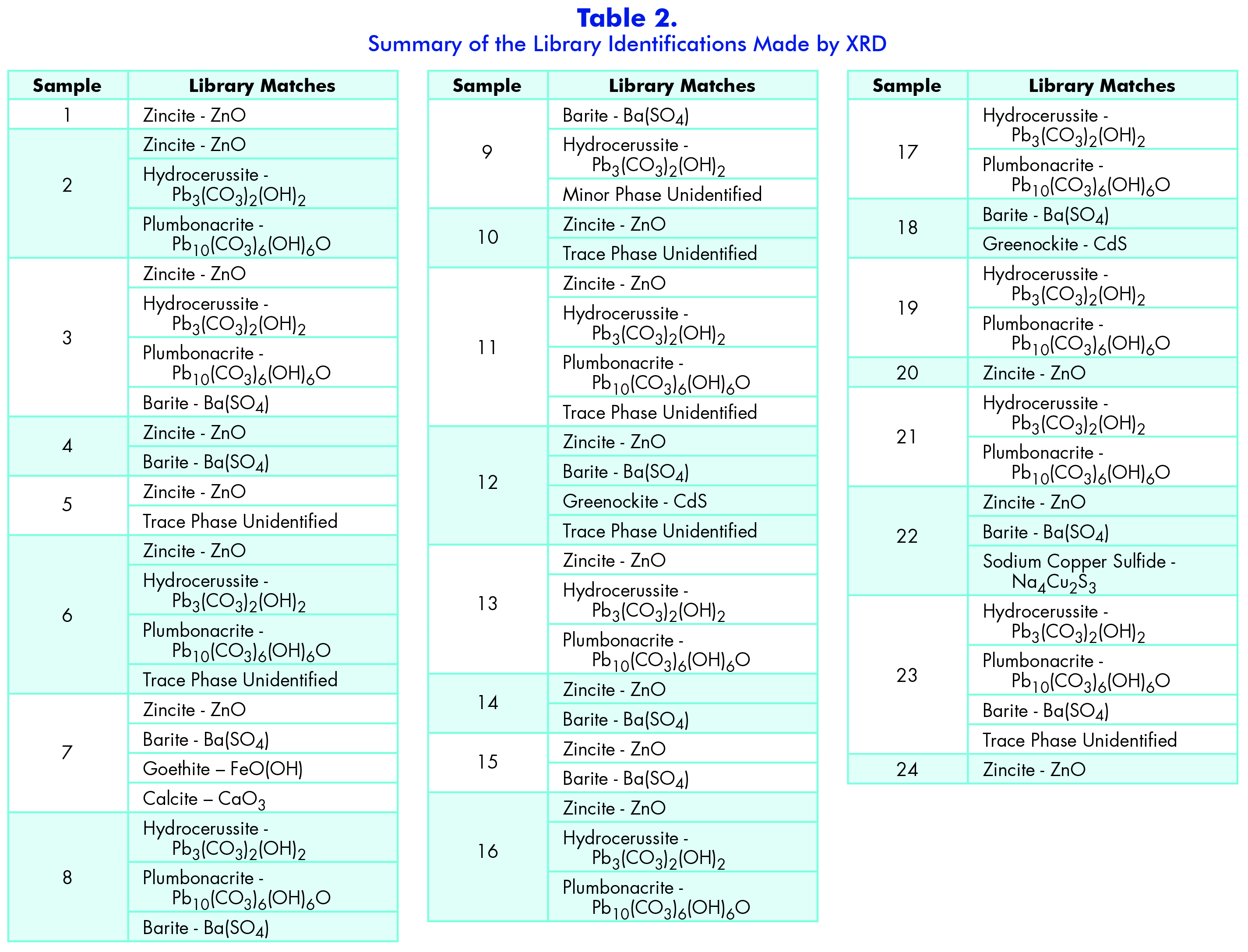
Section 3: Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray Spectroscopy (EDS)
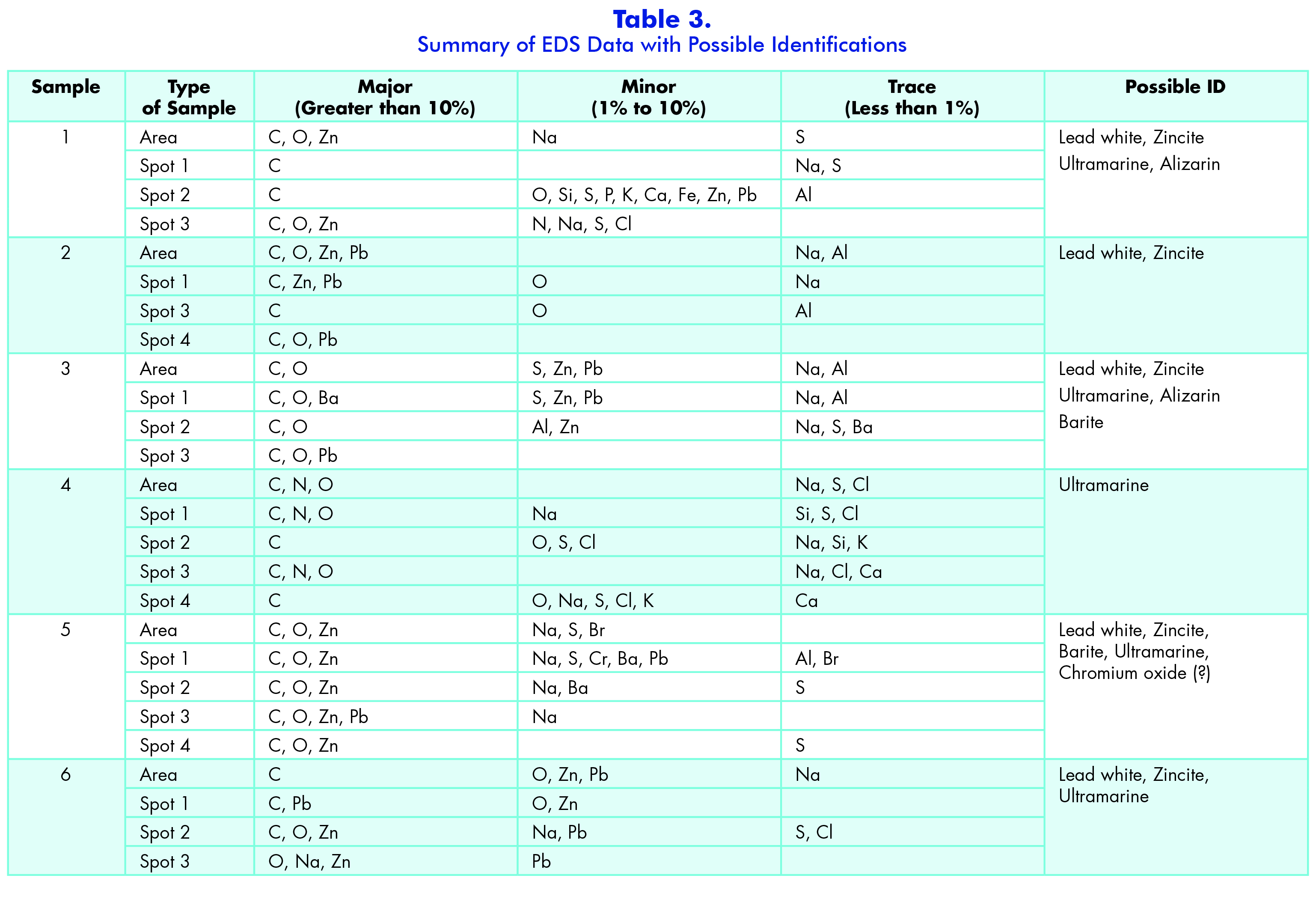
The third instrument I used in my analysis of the paint pigments was a scanning electron microscope (SEM) equipped with an energy dispersive x-ray spectrometer (EDS) detector. An SEM shoots a beam of electrons at a sample and then, depending on depth of penetration, either the secondary electrons or x-rays are detected for imaging or elemental data respectively (Niemeyer et al, 26). The main instrument that I used in my analysis was the JEOL NeoScope benchtop SEM equipped with an EDS detector, specifically a silicon drift detector (SDD). Because it is a benchtop instrument, it does not have the high-resolution or magnification capabilities as a standard low vacuum/variable pressure SEM or a standard vacuum SEM. However, because I was utilizing the SEM for the elemental identification capabilities, it was appropriate.
EDS detectors detect x-rays that are generated by the beam of electrons ionizing atoms in the sample which creates the x-rays specific to elements in the sample. The energy of characteristic x-rays is used for qualitative analysis, i.e. identifying the elements present, while the intensities of the x-ray lines can be used for quantitative analysis or determining the concentrations of the elements. The intensities of the X-rays are affected by the atomic number (Z), absorption (A), and secondary fluorescence (F). Absorption is the biggest factor that affects quantitative analysis since it varies with interaction volume as well as x-ray path length (Niemeyer et al, 83). The SDD used on the NeoScope has many advantages, including high speed, high-count rates, solid state cooling and light element sensitivity. However, one disadvantage of this detector is that there is a high occurrence of artifacts. Artifacts are peaks that appear on the spectra that do not relate to the sample. These can be sum peaks, escape peaks, and silicon fluorescence peaks; however, the artifacts that are most seen with an SDD detector are sum peaks. The others are less common (Niemeyer et al, 89). Due to these artifacts, I had to carefully examine each spectrum I took for each of my samples in order to ensure that all the elements present in my spectra related to my sample.

The sample preparation for this form of analysis was fairly simple in comparison to XRD. Under the stereomicroscope, I placed the MiTegen pin from my XRD analysis on a glass slide on top of a glass block, where I used a medium tungsten needle to remove the particle and soluble gum from the tip of the pin. I then transferred my particle and soluble gum to a carbon planchet where there was a grid scratched into the surface of the planchet. I used this grid to keep coordinates of where each of my samples was placed (Image 3). The soluble gum was necessary to transfer with the particle so that the sample could not move as easily when under the electron beam.
Upon collecting my spectrum, or EDS data, for each sample, I analyzed several spots on each sample preparation and an area of the overall sample. I wanted to get the most representative data to help ensure accuracy. Therefore, for most samples I examined an overall area and three spots on the particle, although some samples had an extra spot or one fewer spot analyzed since the sample appeared to be more homogeneous in nature. Additionally, for these samples I ran them in backscatter electron mode. Backscatter electron imaging (BEI) relies on backscatter electrons which allow me to view relative atomic number or topography I chose the different spots based on the varying intensities of grays because the different grays are due to the different atomic weight or number of varying elements. Heavier elements, like lead, appear lighter or almost white, while lighter elements appear darker. This phenomenon is due to how the electrons interact with different elements. Elements with a low atomic number do not have as many electrons penetrating the sample, which means that not as many electrons are able to leave. Additionally, the interaction volume for low atomic number elements are more teardrop or pear-shaped, which also makes it more difficult for electrons to escape the sample and create a strong signal. High atomic number elements, on the other hand, have more of a hemisphere-shaped interaction volume, which allowed more electrons into the sample and also makes it easier for the incident electrons to escape and create a stronger signal (25).
While I collected the elemental data for each sample, I examined each spectrum for any artifacts that may have been present, such as sum peaks or overlapping energy lines (Appendix C — Part I, Part II, Part III, Part IV, Part V). It is important to note that the software automatically identifies energy lines and peaks based on an algorithm, and therefore it is essential that the analyst review all the results of the software because it is not always correct. In a few of the samples there were some overlapping energy lines that the software mistook and did not correctly identify—for example, Sample 9. As a minor element for spot #1, the computer identified thullium (Tm), which is not a common element in general, but it is also not known to be in paint pigments. Therefore, I used a periodic table with overlapping energy lines and found that aluminum overlaps with thullium and this is a more likely possibility. Aluminum is found in a few red pigments such as alizarin and madder rose. Because this sample is a dark red sample, aluminum is the better option than thullium. Another unusual element that was identified was tungsten (W) in Sample 20. Tungsten’s energy line overlaps with silicon, which is possible since silicates are sometimes used as filler in paints and in the process of creating certain pigments like ultramarine blue. However, another possible source could be from the tungsten needle that I used to prepare the samples. Lastly in a few of the samples including 11, 16, and 19 the element titanium was identified by the computer. It is very possible that the titanium is from the white pigment titanium dioxide; however, titanium dioxide was not seen when examining the samples with PLM, nor did it show up in the Raman analysis. This could mean that it is an overlapping energy peak and a common overlap is barium (Ba), which was seen in a large quantity of the samples. In general, when the barium was present so was sulfur, which means that more than likely barite is present in the samples. Barite is also commonly used as a filler in paints, so it makes sense that it would appear in numerous samples.
Section 4: Fourier Transform Infrared Spectroscopy (FTIR)
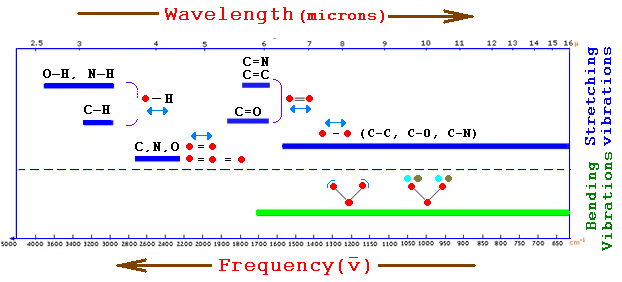
The fourth instrument that I used in my analysis was a micro-FTIR, which is very similar to standard IR, but the sample sizes are considerably smaller. The goal of infrared spectroscopy is to measure how well a sample absorbs different wavelengths of light, in the infrared region. This measurement can be used to identify different molecular functional groups because different groups vibrate at specific wavelengths. Additionally, the energy corresponding to a vibration is specific to different molecular groups. Therefore, if the wavelengths of light absorbed by the sample are measured we can establish a fingerprint of the molecules (Martin and Shearer, 11). Figure 2 below gives the region and vibrational type of common functional groups. In order for a group to be infrared active, a vibration needs to result in a change in the dipole of a molecule. This means that the infrared spectra favor polar molecular groups and asymmetric vibrations since this result in a charge imbalance in the molecule. The intramolecular vibrational energies depend on several factors including mass of the atoms, bond strength, molecular environment, molecular geometry, and hydrogen bonding (Martin and Shearer, 16-17). Because we know the factors that affect the vibrations then we know that certain molecular groups generally fall within a predictable range that can be used to identify the different functional groups. Essentially, infrared spectroscopy follows well-characterized rules, which therefore make spectra predictable.
One of the key aspects in acquiring a good infrared spectrum is sample preparation. For my 24 pigment samples, the sample preparation was fairly simple, especially in comparison to thicker and larger samples. All my sample preparation was done under a stereomicroscope since the sample size was approximately 50 μm. The first step was placing my bulk material slide onto a glass block for easier access. I then used a fine tungsten needle to collect a few particles from the bulk sample which I transferred onto another glass slide where my KBr plate was taped. KBr is the substrate used in the FTIR instruments at McCrone Associates because they are malleable and IR-transparent so they allow for good spectra. Additionally, KBr is easy to cut to specific sizes, so it fits onto FTIR mount easily. Once my sample was placed next to my KBr plate, I picked up a microcover-glass about 3 mm wide with a pair of forceps and placed it over my sample. I used a carbon scribe to press out my sample under the coverglass to make it transparent as possible. Transparency is important for FTIR analysis because it limits the amount of noise in the spectrum. The best illumination for determining if the sample is transparent is transmitted light, since the darker the sample is in transmitted light the more opaque it is. After the sample was pressed out I used my needle to pick up the flattened particles and place them gently on the KBr plate without scratching the surface of the substrate. Then I used the same micro-coverglass and placed it over my sample and pressed it gently on the sample so that the sample was embedded in the KBr. Once the sample was embedded into the substrate, I used my tungsten needle to lightly scribe a box around my sample and I scribed a number above the box in order to easily identify which sample was which during the analysis (Image 4A and 4B).


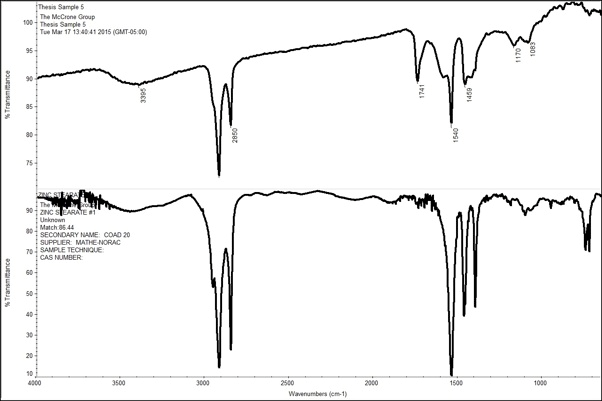
One disadvantage of infrared spectroscopy is that it most sensitive to organic compounds and it has difficulty distinguishing between inorganic compounds. Therefore, many of the 24 samples showed similar spectra. The majority of the samples resulted in a library match to zinc stearate or calcium stearate with another component, meaning it was a mixture. Table 4 identifies key groups that did not match up with the library results. The samples that resulted in a stearate match included 1, 2, 5, 10, 13, 14, 20, and 24. The most likely source of the zinc stearate is the pigment zinc oxide reacting with the fatty acids in the oil-based paint and then forming zinc carboxylates or zinc stearates. This phenomenon is seen most often in deterioration of late nineteenth and twentieth century artwork. The spectrum below shows that zinc stearate is not an exact match for Sample 5; however, it is most likely a component due to the similar peaks at 2900 cm-1, 2850 cm-1, 1540 cm-1, and 1450 cm-1. The peaks around 2900 cm-1 are indicative of aliphatic C-H groups whereas the peak at 1540 cm-1 is indicative of an acid salt. Additionally, the peak at 1741 cm-1 is generally indicative of an ester and that, combined with the aliphatic C-H grouping and the possible C-O group around 1170 cm-1, means there is most likely a fatty component present and, in this case, it is probably the oil in the paint (101-103).
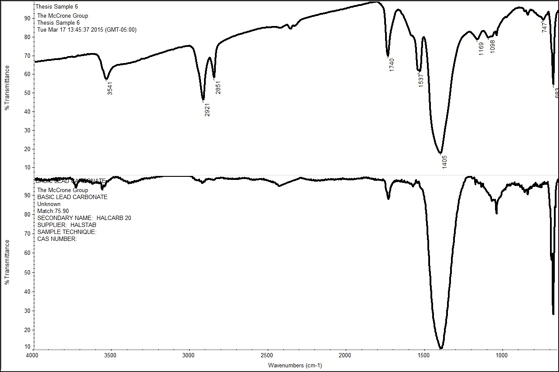
The other major search result match was lead carbonate mixed with another component. This result appeared in samples 3, 6, 11, 12, 15, 16, 18, 19, and 21. Lead carbonate was the best library search match because the strongest peak in the spectrum was around 1405 cm-1, which is indicative of an inorganic carbonates and in this case lead. However, lead carbonate is not the only component present in this sample which can be seen in the discrepancies of the comparison spectra below, for Sample 6. The peak at 1537 cm-1 could be an acid salt like in the previous example, indicating there may be some stearate present. Additionally, the peak at 1740 cm-1 is probably an ester, which is most likely associated with the oil mixed in with the pigments to form the paint.
Sample 9 and Sample 23 were the most distinct out of all the spectra. Both samples were taken from different shades of red on the painting. Sample 9 was a dark red, while Sample 23 was a bright red. Both these spectra have a significant amount of peaks in the fingerprint region (<1500 cm-1) and right above this region as well. A library search for both of them did not yield definitive results, probably because they are mixtures rather than a pure substance. The spectrum for Sample 9 has a peak at 3398 cm-1, which may indicate an amine (NH2) functional group. This spectrum also displays peaks representative of aromatic and aliphatic C-H groups at 3101 cm-1, and 2928 cm-1 respectively. There is also a peak at 1740 cm-1, which, as explained above, represents an ester, which is probably associated with the oil in the paint. The fingerprint region is a bit harder to interpret since there are many peaks, and peaks that meld into other peaks, due to the sample’s heterogeneity. However, there is a large peak at 1408 cm-1 that may be representative of lead. In order to interpret the spectrum further EDS and Raman data is required to narrow down the possibilities. The spectrum for the second brighter red pigment, Sample 23, is slightly different than Sample 9; however, upon closer the inspection there are many similar peaks. It, too, has aromatic and aliphatic C-H groups, as well as an ester group at 1734 cm-1. The main difference between the two spectra is that the spectrum for Sample 23 has more defined and sharp peaks which may be a due to a more concentrated sample or it may be more pure than Sample 9. Overall, FTIR was able to help confirm some of the filler pigments as well as a couple more specific pigments identified with PLM.
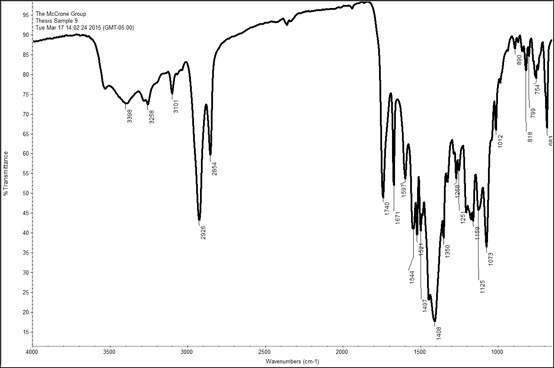
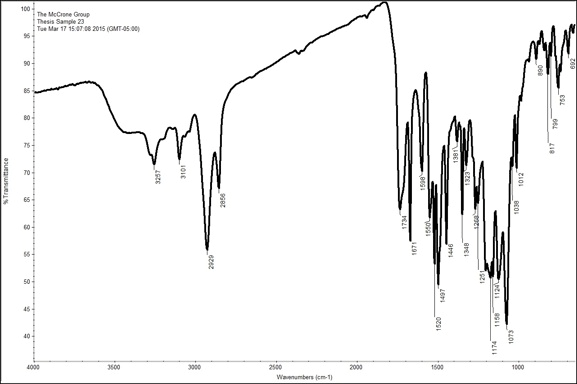
Table 4. Peaks and Possible Functional Groups that Did Not Match Library Result with the Library Result
Raman Spectroscopy Section
The final instrument that I used in my analysis of the paint pigments was a Raman microscope. Raman spectroscopy is very similar to infrared spectroscopy because they both rely on the differing vibrations of specific molecular groups for identification. Also, like in infrared spectroscopy, the intramolecular vibrational energies depend on several factors including, mass of atoms, bond strength, molecular environment, molecular geometry and hydrogen bonding (Martin and Shearer, 15). Despite these similarities, Raman can provide certain data that infrared cannot. The Raman effect occurs when a laser is used to irradiate a sample and the incident beam is inelastically scattered, meaning that it has a different wavelength than that of the original laser. How this process works: a photon of the laser light interacts with an electric dipole of a molecule and excites the molecule to a “virtual” state. The molecule then emits a photon, and most of these photons that are emitted are Rayleigh scatter, but small amounts are inelastically scattered—which is the Raman effect. Therefore, “the difference between the incident energy and the inelastically scattered energy corresponds to a difference in vibrational energy states”(Martin and Shearer, 19). This is different from infrared spectroscopy because infrared uses a broadband IR source and measures infrared light that is transmitted or reflected from a sample, whereas Raman uses visible or near-infrared laser light to excite the sample, and measures the light that is inelastically scattered from the sample (Martin and Shearer, 22). In addition to what is being measured, Raman differs in what molecules show favorable spectra, which is why it complements FTIR. In order for a sample to be Raman active, a vibration must involve a change in the polarizability of the electron cloud, which means that the Raman spectra favor symmetric or non-polar groups. Infrared on the other hand favors polar groups, since it requires a shift in the dipole moment of molecule. Some good Raman candidates are non-polar molecular groups, crystalline materials, polymers, bio-molecules, carbon and oxides.
The sample preparation for Raman is not as meticulous as it is for FTIR. One benefit of Raman is that it can be used to perform in situ examination; however, there are ways to improve the spectra through sample preparation. Good spectra are collected when the sample has an even and flat surface, and is not too sparse. A downfall to Raman spectroscopy is that a sample can be damaged when being tested. This is because the Raman effect is so weak that it needs very high power densities to get results. These high-power densities can result in the degradation or burning of a sample. Additionally, these high-power densities can also cause fluorescence, which is a huge problem when collecting data. This is the main reason that I chose to perform Raman last, so none of my samples would be damaged before I used the other instruments. This also allowed me to use the same sample preparations from my infrared analysis that were prepared on KBr pellets.
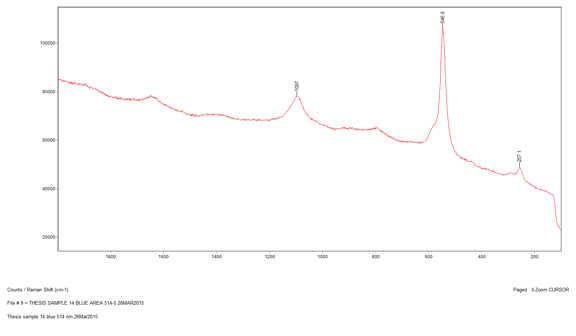
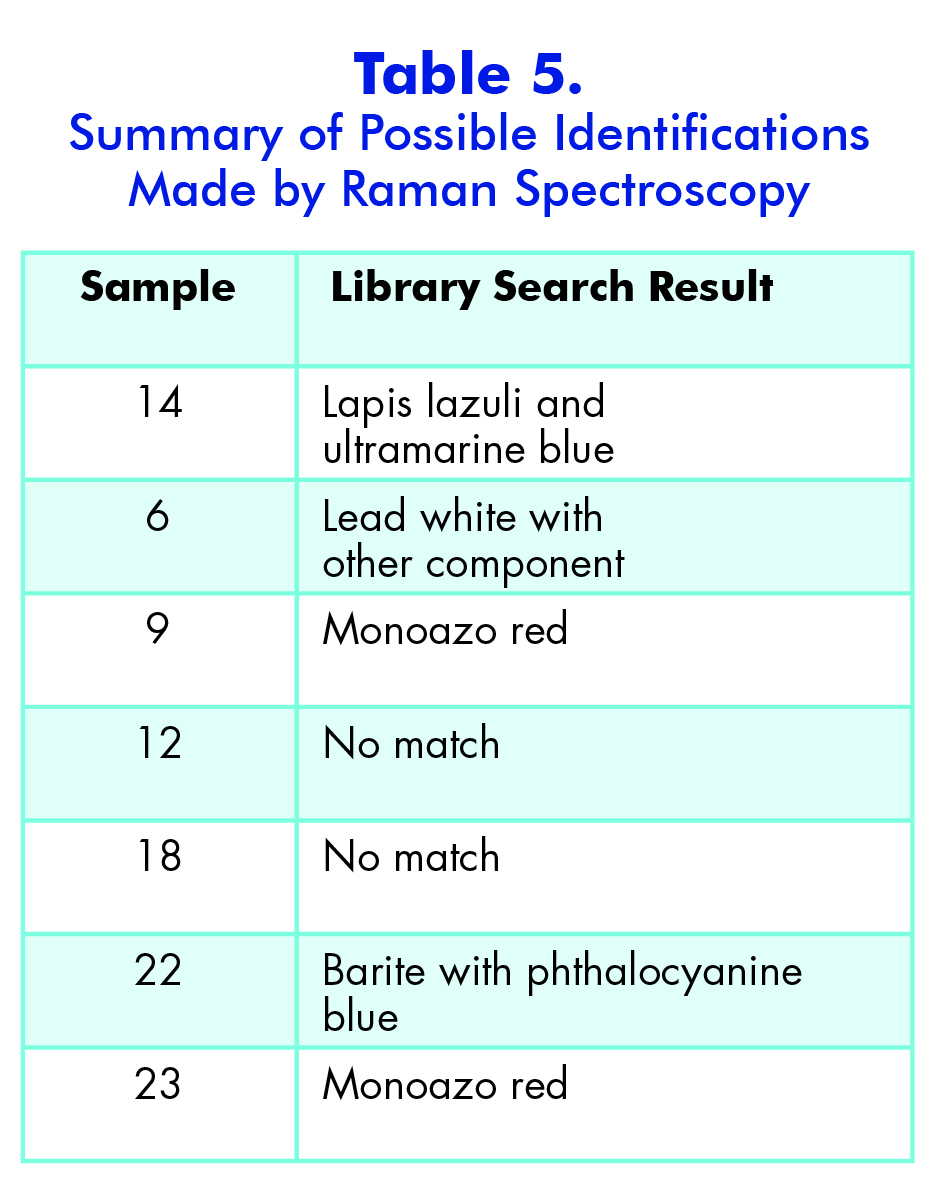
Another reason that I performed the Raman analysis last is because I was able to use the infrared data to determine which samples would most likely work best for Raman. I decided to run seven samples using Raman by analyzing both the EDS and IR data to find which sample identification would most benefit from the Raman. Raman is not sensitive to many functional groups so it would be inefficient to run every sample. The samples that I chose were 6, 9, 12, 14, 18, 22, and 23. All of these samples are slightly more vibrant in color and have slightly different elemental compositions. The first sample that I ran was Sample 14, which was a sky blue sample that I suspected contained ultramarine blue; I wanted to see if Raman could confirm this. I started with the 514-nm laser at 10 percent power, since this is a fairly standard setting for a blue pigment. The 514-nm laser has a higher Raman intensity, which means there is more signal, which in turn means there is higher likelihood of fluorescence. However, in this case I was able to reduce the fluorescence by lowering the power from 10 percent to 5 percent, which allowed me to see a few significant peaks at 1097 cm-1, 546.9 cm-1, and 257.1 cm-1. When I ran a library search of this spectrum through the McCrone Associates database, the best match was lapis lazuli; however, this is a very rare pigment and not generally used in more modern paintings due to cost to process and obtain it, as well as the pigments reaction with a variety of acids which causes it to degrade (Gettens and Stout, 166). The next best match from the search was ultramarine blue, which makes sense because it is the artificial production of lapis lazuli and it is more commonly used since it is easier to obtain and it does not react with acids. However, this reference spectrum does not account for all the peaks in my sample spectrum. The peak that is not accounted for is the one at 1097 cm-1. This may be due to the source of the reference sample or the fact that my sample is a mixture. This peak could represent lead white since it is a common white pigment in paints and the peak is generally seen around 1050 cm-1.
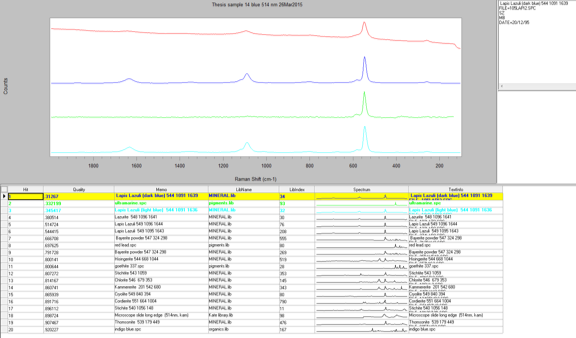
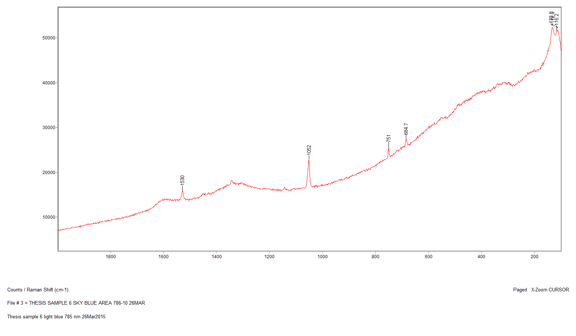
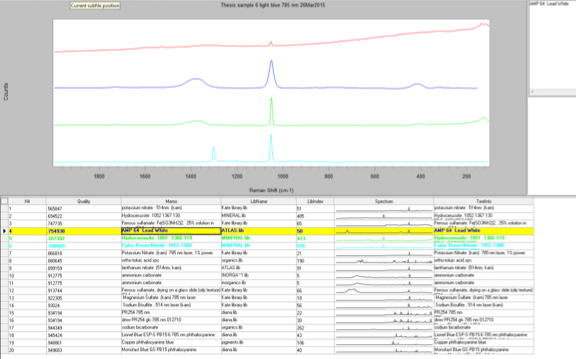
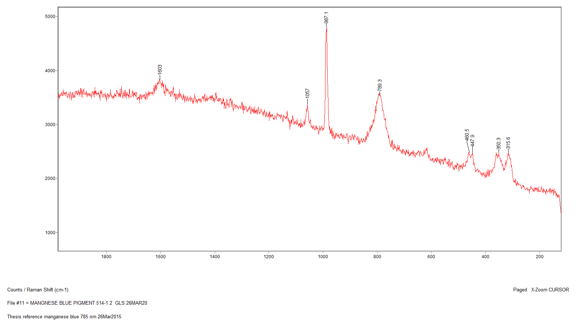
The next sample that I ran was Sample 6, which was the sky blue sample that I suspected contained manganese blue due to my earlier analyses. However, I wanted to see if Raman could confirm this. I began with the 514-nm laser at 10% power but the sample displayed a lot of fluorescence and went off scale. I reduced the power to 0.1% but the sample still fluoresced considerably. Therefore, I switched to the 785-nm laser at 10%, which reduced the Raman intensity so the peaks would not be as strong, but it fixed the fluorescence problem. The peaks that came up were at 1530 cm-1, 1052 cm-1, 751 cm-1, and 684.7 cm-1. However three of peaks were extremely weak, so when I ran a library search, the algorithm found a match to the strongest peak: 1052 cm-1. The best match is lead white, which is consistent with my finding in my other analyses. The strength of the peak could be due to a higher concentration of the lead white than the other components in the paint, or because lead white is the best Raman scatterer out of all the components. Additionally, the library did not have a reference spectrum for manganese blue, so I prepared a sample of known manganese blue mixed with phthalocyanine blue to see if any of the peaks were similar. It is difficult to say if certain peaks line up since both samples are mixtures and the spectrum could be slightly different due to differing hydration. The reason that I used a mixture for my reference sample of manganese blue is because it was the only known manganese blue sample that I had available to me, so in the future I would like to get pure sample and compare it again with my sample.
The next sample that I ran in the Raman microscope was a dark red sample, Sample 9. This Raman spectrum had many peaks that were fairly strong which helped get a more definitive match than in the previous samples. The sample produced the best results with a 785-nm laser at 10% power. The library search yielded many results for different types of monoazo red pigments. Therefore, the sample is most likely some version of a monoazo red.
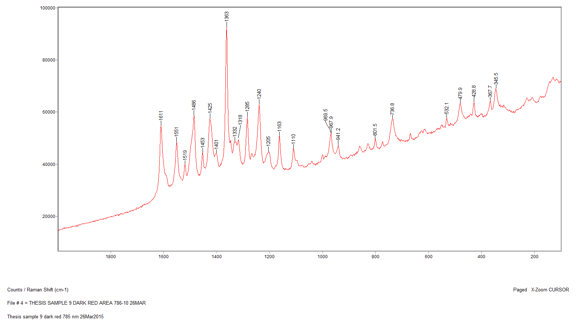

Sample 12, a pinkish red section was run after Sample 9. I chose to run this sample in hopes of confirming the presence of cadmium red since this was probably a pigment that was present according to PLM and EDS data. However, the Raman spectrum for this sample was inconclusive. There were two varying shades of pink/red in my sample preparation, so I gathered a spectrum for each of these sections. The spectrum of the first darker pink section was very noisy and contained only two definitive peaks at 1609 cm-1 and 1363 cm-1. The second lighter pink section had these two peaks plus three more between them at 1552 cm-1, 1489 cm-1, and 1422 cm-1(Appendix E). I ran a library search for both spectra but neither produced any reliable results. This could be because the library does not have the proper reference spectrum for this pigment or the fact that it is a mixture is not allowing for more conclusive results. Therefore, in order to get an accurate identification of this sample I will have to compare all my data from the different instruments to reach a conclusion.
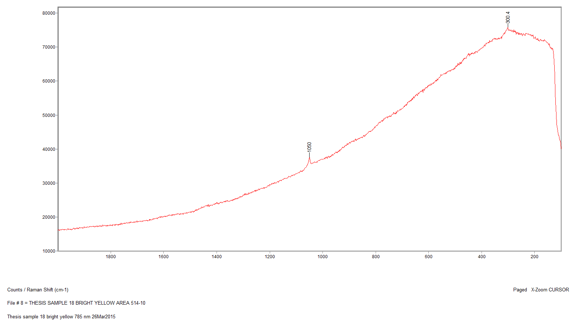
I then analyzed the bright yellow sample from the painting, Sample 18. I found this sample was extremely beam sensitive and burned under the laser beam. The best conditions that produced a peak on the spectrum were using the 510-nm laser at 10 percent power. Nevertheless, the spectrum displayed only one peak at 1050 cm-1, which is most likely due to lead white. This is may be due to the yellow pigment not being a good Raman scatterer.
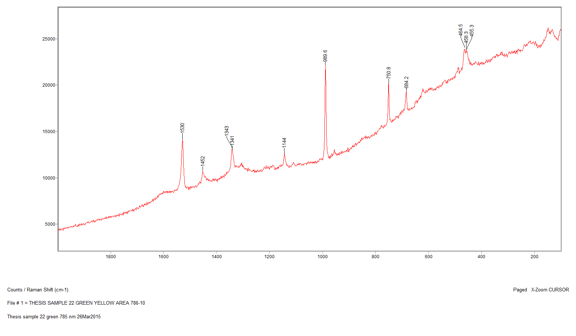
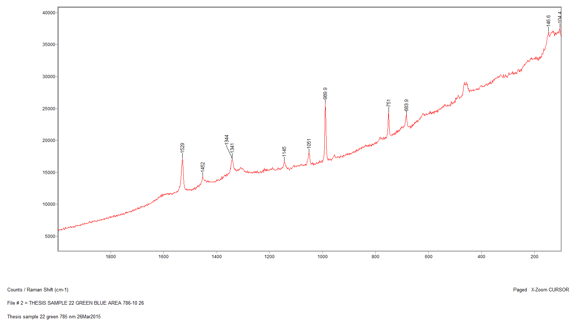
The penultimate sample that I analyzed was Sample 22, a bluish green section. When I looked at the sample through the microscope I saw two distinct colors: greenish yellow and greenish blue. The first section I analyzed was the greenish yellow section. I used the 785-nm laser at 10 percent power and was able to gather an acceptable spectrum with not too much noise. There were several peaks on this spectrum ranging from 455 cm-1 to 1530 cm-1. However, when I conducted a library search for the spectrum, the search algorithm focused on the strongest peak at 989.6 cm-1, which indicates there is barite present. Furthermore, because the barite reference spectrum does not account for all the peaks, it is most likely a component of the paint and not representative of the whole. Then, I ran the greenish blue section and came up with a very similar spectrum. I surveyed the different library search results to see if any of them could explain the peaks that did not match up with barite. The best reference I could find was for a phthalocyanine blue pigment. This is a possibility because the EDS data showed that there was copper present in the sample, which is used to create phthalocyanine blue.

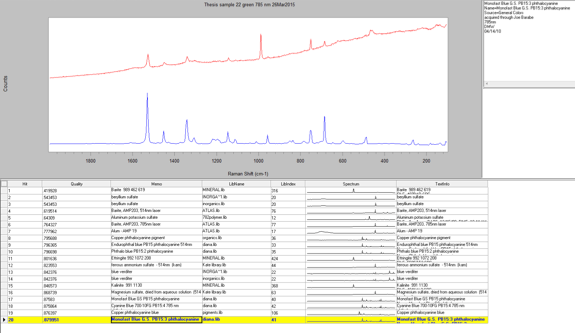
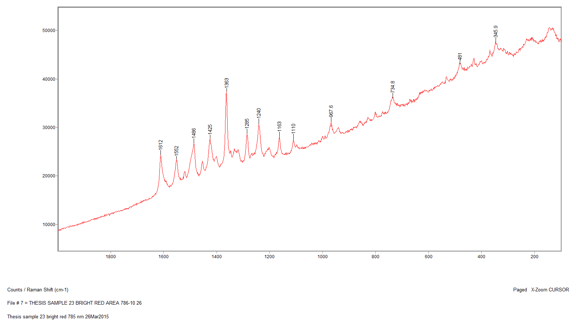
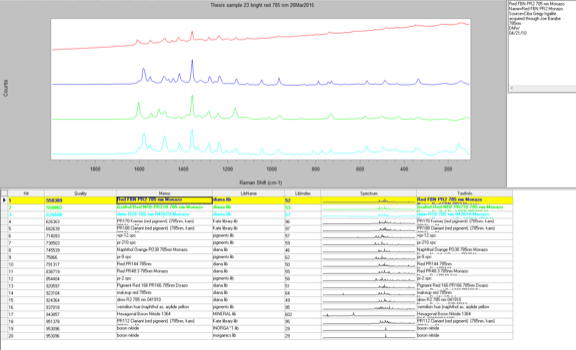
The last sample that I chose to analyze using the Raman microscope was a bright red sample, Sample 23. This sample appeared uniform in color when examined using the microscope so only one spot was analyzed. This spectrum contained many peaks, both strong and weak. It looked very similar to the other red sample I ran earlier, Sample 9. When I conducted a library search the results were exactly the same as Sample 9. This means that this red pigment also must be a version of the monoazo red pigment and since it was taken from the same painting as the previous sample it is most likely the same pigment.
Results and Discussion
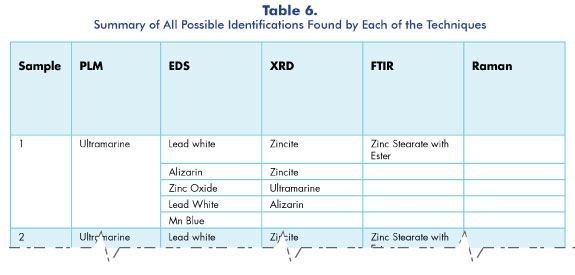
From Table 6, it is clear to see that the most prevalent pigments were lead white, barite, and zinc white. This is not too surprising since these are often seen as either fillers or bases in many paintings. Barite, for instance, is commonly used for a base in lake pigments. Additionally, in judicious quantities it may improve the wearing and weathering qualities of zinc and lead white, which further supports why so much of the three compounds were seen together (Gettens and Stout 96). Zinc white began to be commercially sold in 1834, however artists noticed that it often became dry and brittle, so many of the artists combined lead and zinc white to see if they could counteract these affects (Gettens and Stout 178).
Many of the pigment types only appeared in one or two forms of analysis. One such pigment type were the two cadmium pigments: cadmium yellow and cadmium red. These pigments were seen using PLM, however they were difficult to identify using only this method due to the abnormal morphology. Normally, cadmium pigments are 1 to 2 μm, but the pigments I identified were much bigger, anywhere from 5 to 12 μm. However, the other PLM characteristics were a match for the cadmium pigments. Additionally, in a few of the samples, once they were adequately dispersed, I saw pigments that were more consistent with cadmium red and yellow. The EDS analysis supported the PLM data because it showed that there was cadmium and sulfur in several samples along with cadmium and selenium. It was because of this data that I decided to go back to my PLM samples and redisperse my pigments. One concern I had was that some of the cadmium reds seem to be slightly different hues; however upon further research I discovered that cadmium reds can slightly different in hues due to the proportions of cadmium to sulfur or selenium. The cadmium yellow can also vary in color from lemon yellow to orange due to crystallinity of the sulfide (Gettens and Stout, 102). Also, in two of the XRD samples I did detect cadmium sulfide, which further supported the presence of cadmium pigments.
Another pigment that was not observed in all the analytical methods was manganese blue. This pigment was only seen using PLM, but it was quite distinguishable. This pigment is not generally observed with EDS because there is such a small amount of the manganese used to make the pigment. Furthermore, the way it is produced is with a barium manganite that is fixed on a barium sulfate base, which could quite possibly be another source of the barium seen in the EDS data. Manganese blue is quite distinguishable using PLM because it is pleochroic, has a unique shade of blue, distinct morphology, and is anisotropic. This pigment is also useful in determining approximately when this painting was painted because this is a relatively new pigment and it was not introduced until 1935, so the painting would have had to been painted after that.
One other pigment that was seen only with one technique was the monoazo red. This pigment was only identified using the Raman microscope; however, it was noted in the PLM section that there was a red that appeared different than the others, but it could not be identified. Azo compounds are often used as dyes for paints in the yellow to red range. They are formed from diamide compounds, which may be why Raman was able to detect it and the other techniques could not. Raman is more sensitive to N=N bonds since they are symmetrical and non-polar.
In order to improve this experiment in the future, I think it would be beneficial to isolate specific pigments by color and morphology, so I could obtain more data on the less abundant pigments rather than the fillers. I also think that using more than one particle with longer counting times for the EDS analysis would be beneficial and it might help get a more representative sample. Overall, this study accurately demonstrated how each of these techniques could be used to identify different pigments. This study also demonstrated that each technique has its limitations and strengths and together the different forms of analysis can work together to compensate for these weaknesses. For instance, PLM can be used to determine morphological characteristics, but pigments tend to be very small so it is difficult to characterize. XRD is able to identify crystalline structure, which in turn can lead to an identification. However, if a material is not crystalline, XRD is unable to conclusively identify it. SEM-EDS is able to give the elemental data of a pigment, but not the compound information, for compound information XRD is a useful technique. FTIR can also be complemented with SEM-EDS and XRD because it is most sensitive to organic compounds, and therefore does not provide the whole picture on its own. Lastly, Raman supplements FTIR because it can pick up the inorganic compounds, however it can damage the sample, which is why this technique should be performed last. In conclusion, these techniques alone cannot provide a positive identification, however, working together they can give a more complete analysis and identification.
{kind=link}
Bibliography
Barabe, Joseph. Pigment Identification. Westmont, Illinois: Hooke College of Applied Sciences, 2014. Print.
Caple, Chris. Conservation Skills: Judgment, Method and Decision Making. New York: Routledge, 2000. Web.
Gettens, Rutherford J., and George L. Stout. Painting Materials: A Short Encyclopaedia. New York: Dover Publications, 1966. Print.
Martin, Kate, and Gretchen Shearer. Infrared Spectroscopy. Westmont, Illinois: Hooke College of Applied Sciences, 2014. Print.
Martin, Kate, and Gretchen Shearer. Raman Spectroscopy. Westmont, Illinois: Hooke College of Applied Sciences, 2013. Print
“McCrone Atlas of Microscopic Particles.” McCrone Atlas of Microscopic Particles. The McCrone Group, 01 Apr. 2005. Web. 17 Feb. 2015.
Niemeyer, Wayne D., Rebstock, Joseph M., and Craig S. Schwandt. Scanning Electron Microscopy. Westmont, Illinois: Hooke College of Applied Sciences, 2014. Print
Reusch, William. “Infrared Spectroscopy.” Infrared Spectroscopy. 05 May 2013. Web. 08 May 2015.
Sotiropoulou, Sophia, and Sister Danilia. “Material Aspects of Icons. A Review on Physicochemical Studies of Greek Icons.” Accounts of Chemical Research 43.6 (2010): 877-87
Stoner, Joyce Hill. “Changing Approaches in Art Conservation: 1925 to the Present” Scientific Examination of Art: Modern Techniques in Conservation and Analysis (2003): 41-57
Van Howe, Tom. Polarized Light Microscopy. Westmont, Illinois: Hooke College of Applied Sciences, 2013. Print.
Comments
add comment