X-ray Diffraction–Solving Problems with Phase Analysis
July 28, 2017
Presented by: Joe Swider, Ph.D.
The difference between two phases of the same composition can mean the difference between useful or toxic. This is true of materials including pharmaceuticals and food additives. X-ray diffraction (XRD), in use for more than 100 years, can quickly distinguish between crystalline phases of a wide variety of materials such as active pharmaceutical ingredients, paints and pigments, and corrosion. 28 minutes.
Download the slides.
(Note: due to animations within the presentation, some images and captions overlap in the PDF format; you may view the animations in the video, or see many of the hidden images below, and in the downloadable transcript.)
Transcript
Charles Zona (CZ): Hi, my name is Charles Zona, and I’d like to welcome everyone to today’s McCrone Group webinar. Our presenter is Joe Swider of McCrone Associates. Joe is going to talk to us about solving phase analysis problems using X-ray diffraction. Joe will field questions from the audience immediately following today’s presentation, and now, I will hand the program over to Joe.
Joe Swider (JS): Hello, and good afternoon. Welcome to the McCrone Associates webinar “X-ray Diffraction: Solving Problems with Phase Analysis.” I’m Joe Swider, a senior research scientist at McCrone. I have been with McCrone for almost 14 years in their electron optics group. I routinely look at samples using X-ray diffraction, and also scanning electron microscopy with energy dispersive X-ray spectrometry, or SEM/EDS.
Today, I’m going to talk about how McCrone uses X-ray diffraction to solve many materials analysis problems. I am going to go through a small amount of background for X-ray diffraction, show you what kind of instrumentation and capabilities we have at McCrone Associates, and then illustrate that instrumentation with some case studies.
We can start with the basics. Phase identification of materials, simply explained: different phases of materials with the same elemental composition can have extremely different properties. X-ray diffraction can distinguish between these different phases. It’s commonly used for inorganics, but more and more is being used for organics, in particular, with pharmaceuticals. You can also distinguish the degree, or if, something is crystalline or amorphous using x-ray diffraction.
How is phase identification different than typical elemental identification? In elemental identification the question is, what is the amount of each element in the sample? For instance, on this slide, I have titanium dioxide. The mass percent stays the same no matter how those atoms are arranged; however, titanium dioxide can exist as at least three different forms. You can see, on the right side, the phase. How the atoms are arranged in a sample can show very different physical properties, if you look at rutile, anatase, or brookite. Another good example is calcium carbonate, a very common material. You can see, on this slide, an image of calcite and an image of aragonite. Both of the samples, if they were analyzed using EDS or some other elemental technique, would show exactly the same amounts of calcium, carbon, and oxygen; however, you can see—because the atoms are arranged differently—they look very different. Property-wise they are very different. Calcite, commonly known as chalk, is very different than aragonite, which is known to be a portion, or part of, mother-of-pearl.
Now I will briefly explain a small amount of X-ray diffraction theory. X-rays interact with materials as waves, and when they do this, they can interfere with each other, depending on the phase of the wave. If they interact constructively, it produces a bright line in a diffraction pattern. If they are out of phase, they actually interfere destructively, and no bright reflection appears on the X-ray diffraction pattern, but a bright reflection will occur when the half-life difference is an integral number that satisfies the equation here, nλ = 2d sin θ, which we call Bragg’s Law. I would like to point out that this is a very simple schematic on here, but X-ray diffraction, because those waves interact specifically with the atom and where it is in the crystal lattice, that it produces a very specific XRD pattern. This is why X-ray diffraction is very useful for phase identification.
Typically, we look at samples that are called powder samples. With single crystals—a single crystal is where you have a sample and the rows, columns, and all atoms are in an order that is unchanged throughout the whole crystal no matter how large or small it was—that’s typically a single crystal. But in real life, we typically have powder samples. Most times, the crystals are formed at angles to each other, or we have, maybe, another crystallite that is at a different angle to all the other particles or crystallites in the sample.

What happens is, they all bunch up, and we obtain what is known as a powder sample. There is actually an advantage to this. When we use X-ray instrumentation, which is pretty simple actually, we have an X-ray source and a sample, and the X-ray source will aim the X-rays at the sample; the sample must rotate, and by rotating the sample, we expose all the different planes of all the different angles in those little crystallites in the powder sample. As the sample rotates, it does give off constructive interference, which are bright lines in a pattern and are detected. They can be detected electronically through a charge coupled device (a type of camera), or they can be collected all at once on film. The instrumentation we use at McCrone Associates uses a filmless film technique.
This is an example of an XRD ring pattern, which many of you, if you’ve done XRD decades ago with photographic film, it looks about the same, or may be a little wider. The center of that circle, where there is an “x,” is actually where the sample would be, and the diffraction pattern rings are outward from that center. We use system software to translate the ring pattern to a 2θ versus intensity plot such as this. You can see the x-axis is 2θ degrees; the y-axis is intensity.
I would like to point out that it is translating this directly from the image, so, if you see on this ring pattern, if you start from the center where that “x” is, and let’s say that is the “zero,” and move anywhere out in the ring to increase your 2θ angle, you can see the first three rings you hit are very intense; some space; a less intense ring; and then a more intense ring. If you look at our XRD data, you can see that the first three peaks going from left to right between 20-30 are very intense, which of those three rings a smaller peak, which was the weaker ring, etc.

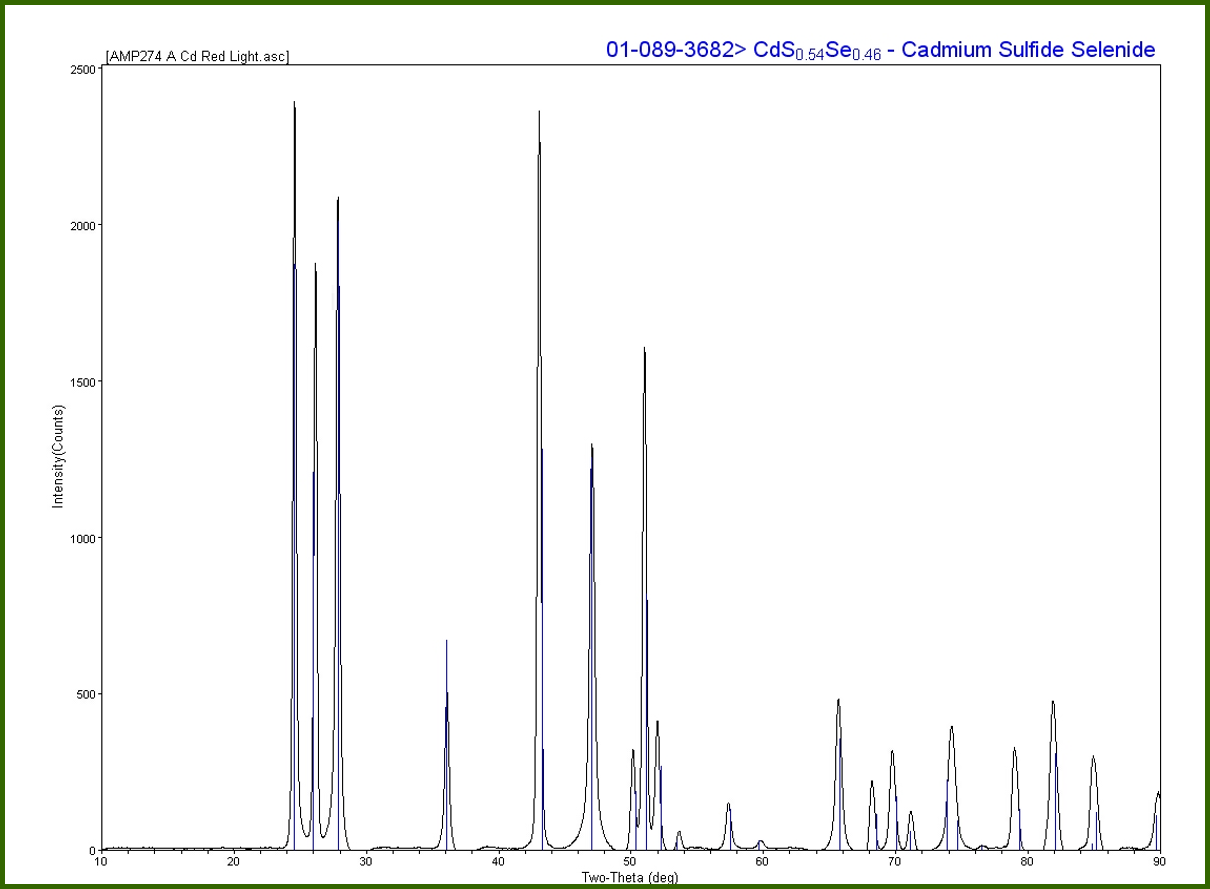
We need to translate the XRD data from the image to the 2θ versus intensity for reference purposes. Almost all references by the International Center for Diffraction Data (ICDD), which is a database we use, is in the form of 2θ versus intensity. The ICDD database is updated every year, and it has over 350,000 references.
I would like to talk about the instrumentation we use at McCrone Associates for XRD. We have two Rigaku micro XRD instruments, both instruments utilize a rotating anode, which is a high intensity x-ray source; much higher than typical sealed tubes. The higher source enables us to provide clients with a much faster turnaround time for analysis, and analysis of much smaller particles.

We also use an imaging plate for detection, which is the image you saw earlier—the ring pattern was used with this imaging plate, which is in the image here as the curved structure behind the sample. Particles can be in hundred microns down to 10 µm in size, and the beam size for these both of the instruments is 100 µm.
Now that we have this instrument, we utilize our exceptional cleanroom for extraction and mounting particles. Our cleanroom can extract particles for various techniques, but also can mount them for XRD. For XRD, for particles, the cleanroom can draw out fiber optics to the size of about 5–10 µm for the mounting of small particles.
Here’s an image of a tungsten needle mounting a particle onto one of these thin glass fibers. The SEM image shows this more explicitly with a 9 µm particle adhered to the glass fiber, just using a simple adhesive.


Now that these particles are mounted and we know how they are mounted, how would this fit into an analysis scheme? There are a couple of ways we can do it. Typically, particles are analyzed for SEM/EDS analysis first. We get elemental information, and imaging, or morphological, information. Then, if the project leader or the client decides we need phase analysis, the sample is brought back to the cleanroom, mounted for XRD, and analyzed. This mount that I’m showing you here is a polyimide mount, or kapton. It is very low absorbance of X-rays and flexible, and can actually support much larger particles than the fibers.
Typical XRD analysis is done, translated 2θ versus intensity, and compared to a reference. Alternatively, the cleanroom can extract the particle and directly mount it for XRD; we can perform the XRD analysis, obtain the XRD data, and then remove it from the XRD mount for SEM/EDS if we feel we need the elemental or morphological information, but these are mostly for particles.
We have many samples that are powders. Powder samples are usually packed into a glass capillary. The glass has a low absorbance for X-rays and is not crystalline, so it doesn’t interfere the X-ray pattern. You can see in the middle image, these capillaries are about 100 µm inner-diameter. We use a 100 µm beam, so the amount of sample we require is very little. We can also look at surfaces or areas on large samples as pictured on the right.


With particles, powders, and bulk, these are some of the types of samples we receive for analysis. We can also work with particles that are extracted, or as a result of filtration, isolate them and analyze them on SEM/EDS and/or XRD. Usually, when something comes in for XRD, we will ask if you need elemental analysis, and what kind of data analysis do you need? Do you need a simple comparison to the ICDD? Do you need to know if it’s crystalline or amorphous? Or, do you have your own standard to compare to? Some common requests we get for analysis are pharmaceutical verification of phase. We can also file USP 941, which is the pharmaceutical method for X-ray diffraction. We look at contamination identification, corrosion products, paints and pigments, a wide variety of organics, and can determine crystallinity.
Some limitations we have are quantitative XRD and percent crystallinity. We can do this on a limited basis, and on a client-by-client case. Quantitative methods with X-ray diffraction are usually good around 5–10% uncertainty. Some of the things we are not set up to do are determining the crystal structure of an unknown, and this is also known as indexing or single crystalline analysis. We also do not have cryo or low temperature capabilities, which is more in tune with protein analysis. We cannot look at in situ samples that are very large.
Do you have a materials issue? Speak with a scientist.
I would like to provide some examples of XRD analysis that we’ve come in contact with at McCrone. The sample that you have in front of you shows a pattern with three different types of blue pigments all mixed together in the same sample; however, the XRD pattern clearly reveals that there are three different types of pigments. Using elemental analysis, you may see all the elements from all three pigments, but you might not be certain which ones are there. You can see, XRD clearly shows that these three phases are present.
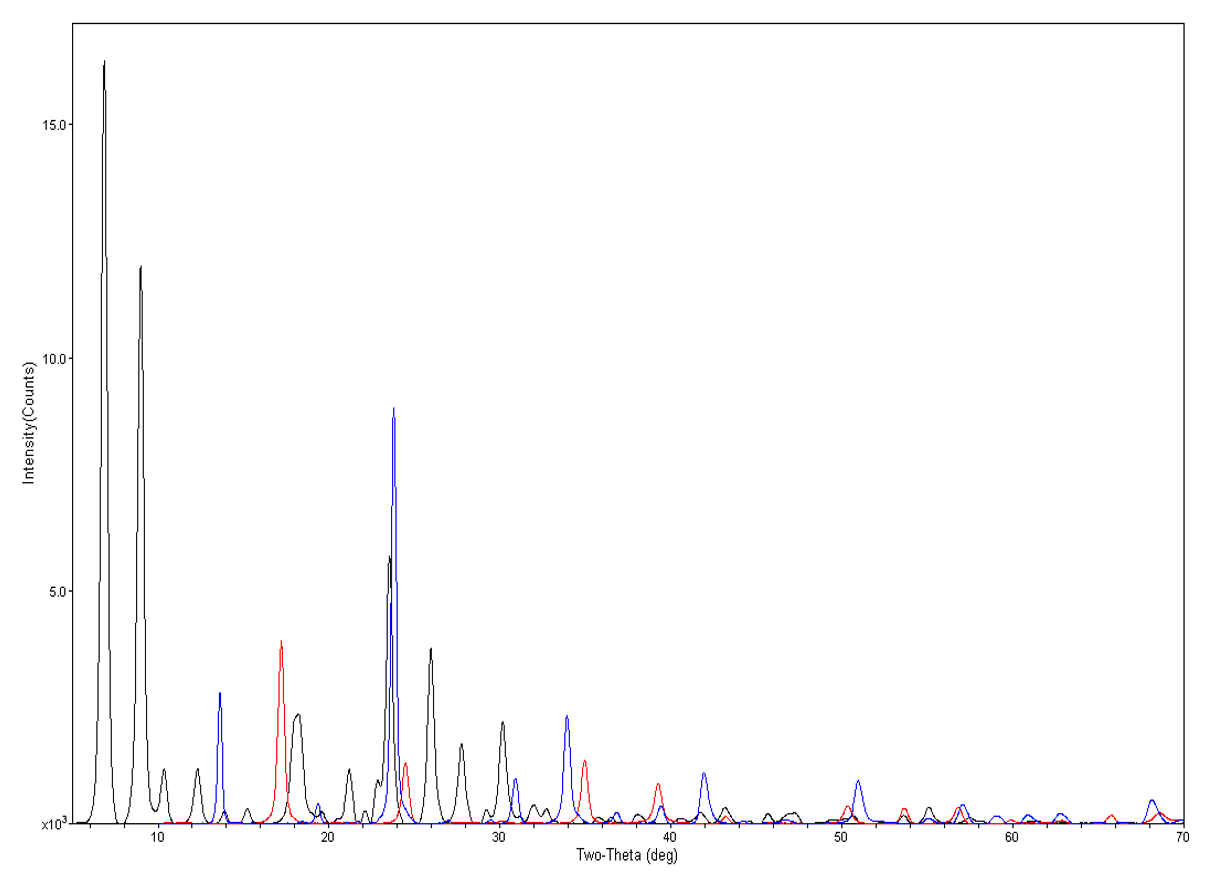
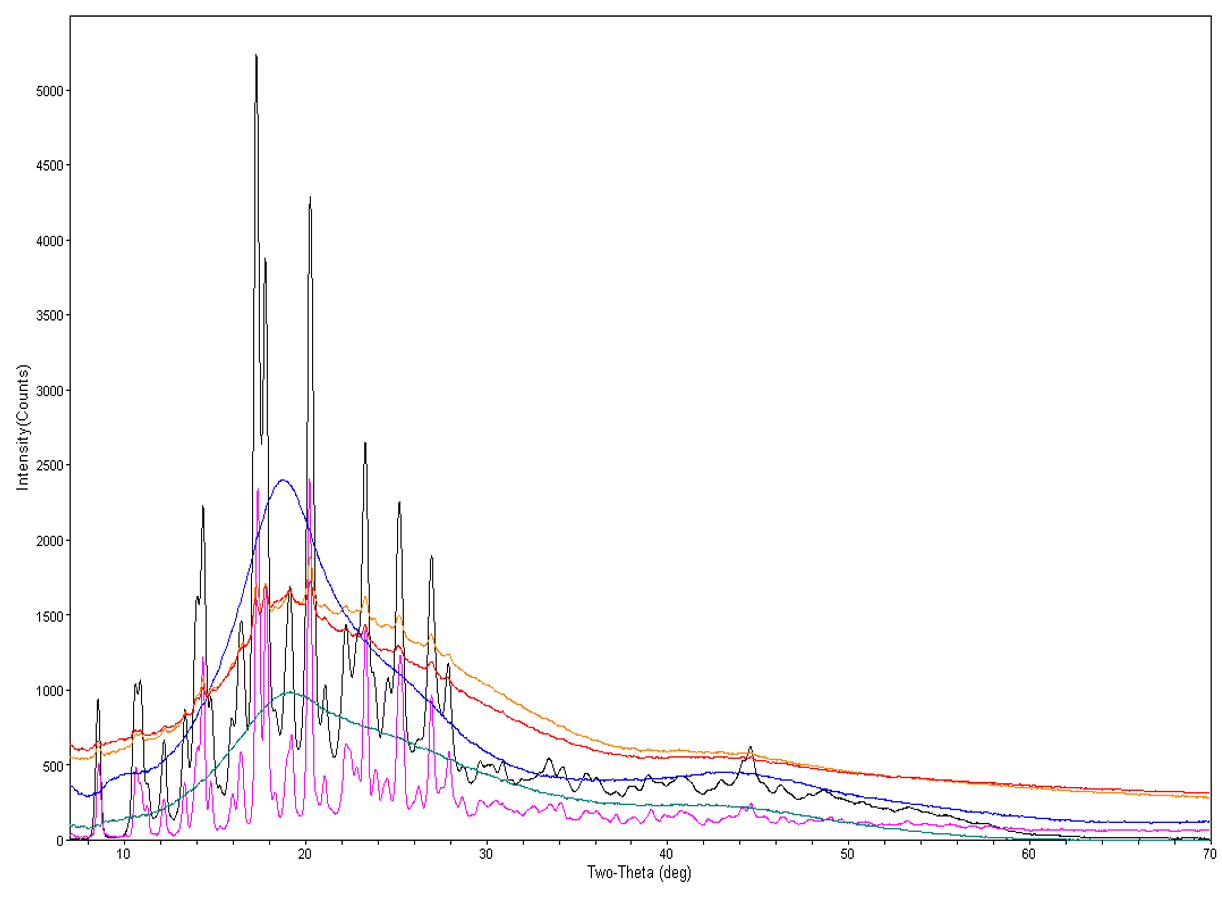
We mentioned crystallinity before, now the crystalline pattern is very very sharp, sharp peaks—where the amorphous pattern is very amorphous, what we call a hump, which is just scatter from everywhere because there is no order to the crystal structure. We can also see in between there is a semi-crystalline material. This material in question is crystalline, and then when heated, becomes amorphous. These are different stages of heating for this type of material.
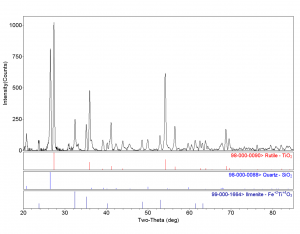
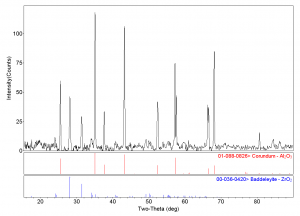
Here are some particle examples. This is a 20 µm particle that we analyzed and was revealed to have three different mineral phases: rutile, quartz, and ilmenite. If you look at this particle using SEM/EDS, I doubt you’d be able to tell those three phases were definitely there. Likewise, with a 10 µm mineral, if you had analyzed this, you would find zirconium, oxygen, and aluminum. We wouldn’t know what type of mineral it was, when, in fact, it’s actually corundum and zirconium oxide.
(Click on the images below to enlarge.)
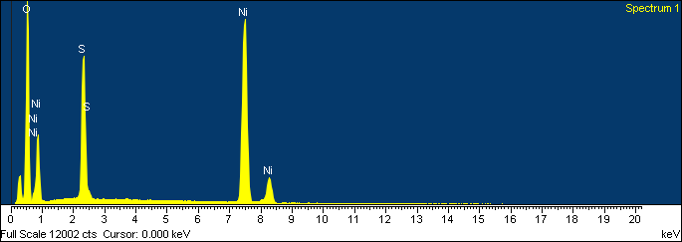
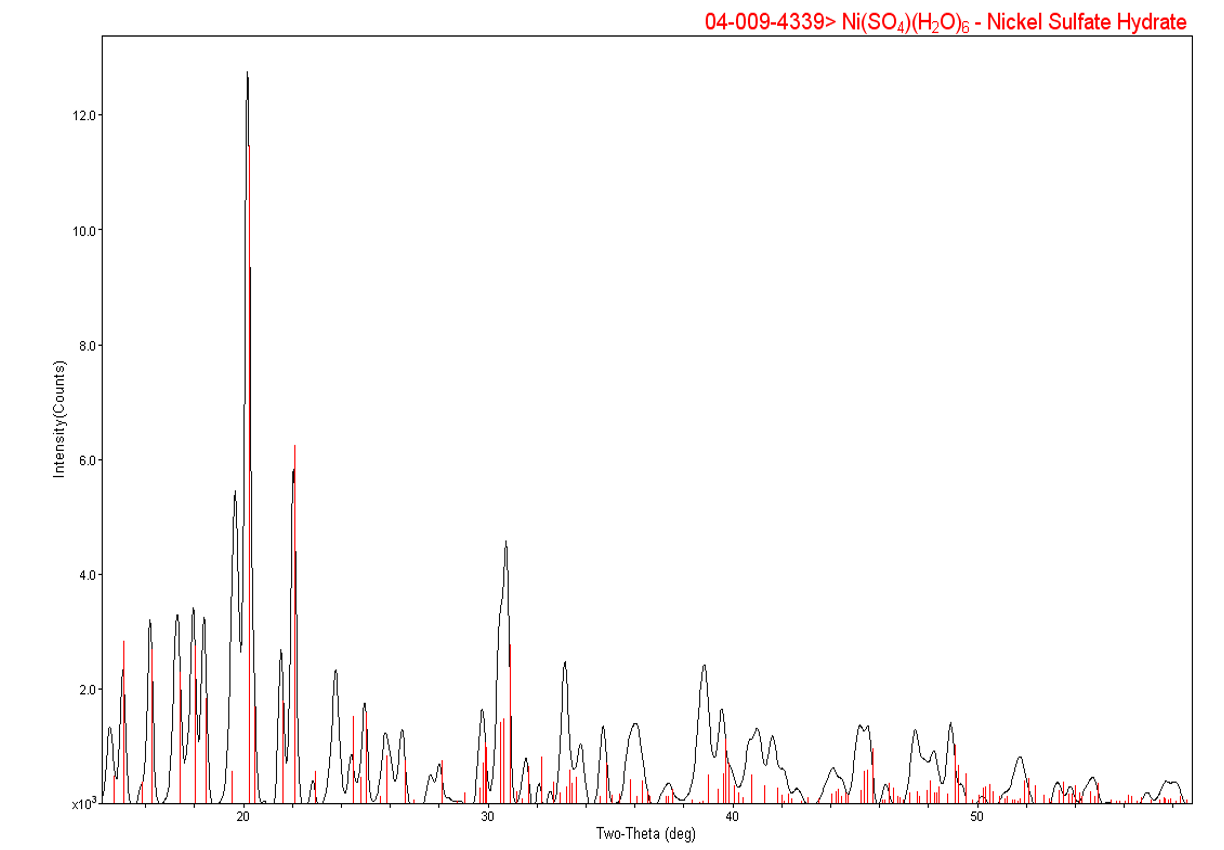
A practical example we’ve done is corrosion identification. We received this sample from a corroded circuit board, and removed some particles from the circuit board. SEM/EDS showed the particles were mostly nickel, which was the base metal on the circuit board, but lots of sulfur. Sulfur is known to be a corrosive agent, but the client was more interested in exactly what phase was there. The sample was then removed from the SEM/EDS mount, mounted for X-ray diffraction, and shown to be nickel sulfate hydrate. This something you may not see by SEM/EDS, because they cannot detect hydrogen by that method.
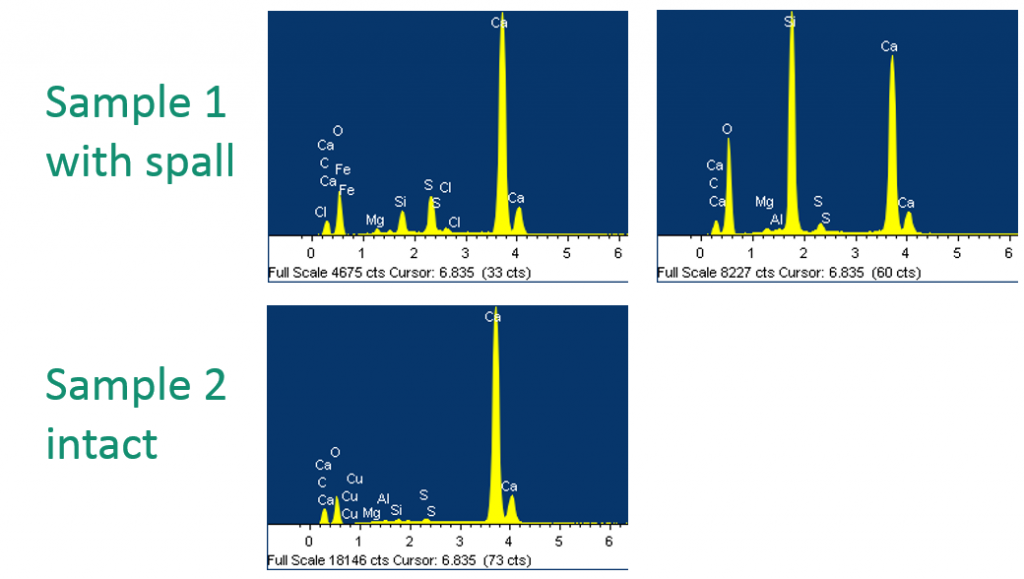
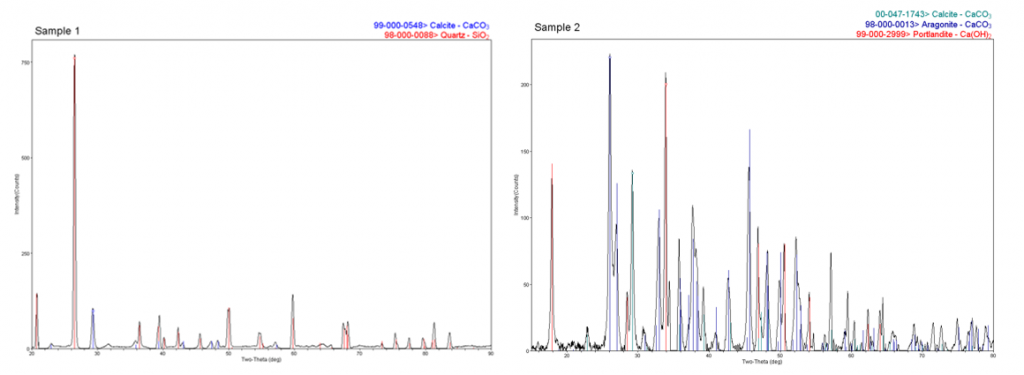
A case study for some other particles was fresco deterioration. Several frescoes in a university hall were showing spall, or accretions, on their surface. The conservators wanted to know what was the composition of the spall and the composition of the intact material, so samples were taken from both. You can see here—SEM/EDS was done first—the sample that had the spall was mostly calcium, which is pretty expected with a fresco, but also large amounts of silicon, which was rather unexpected. The intact sample was mostly calcium, oxygen, and carbon, with small amounts of trace elements. If we analyze these by X-ray, you can see Sample 1 did, in fact, have a lot of quartz in it—actually, a large amount of quartz in the sample, in addition to calcite; whereas the non-spall sample was a combination of calcite, aragonite, and calcium hydroxide or portlandite. This gave the conservators a lot of information to distinguish where the spall was coming from, and what the original fresco was made of.
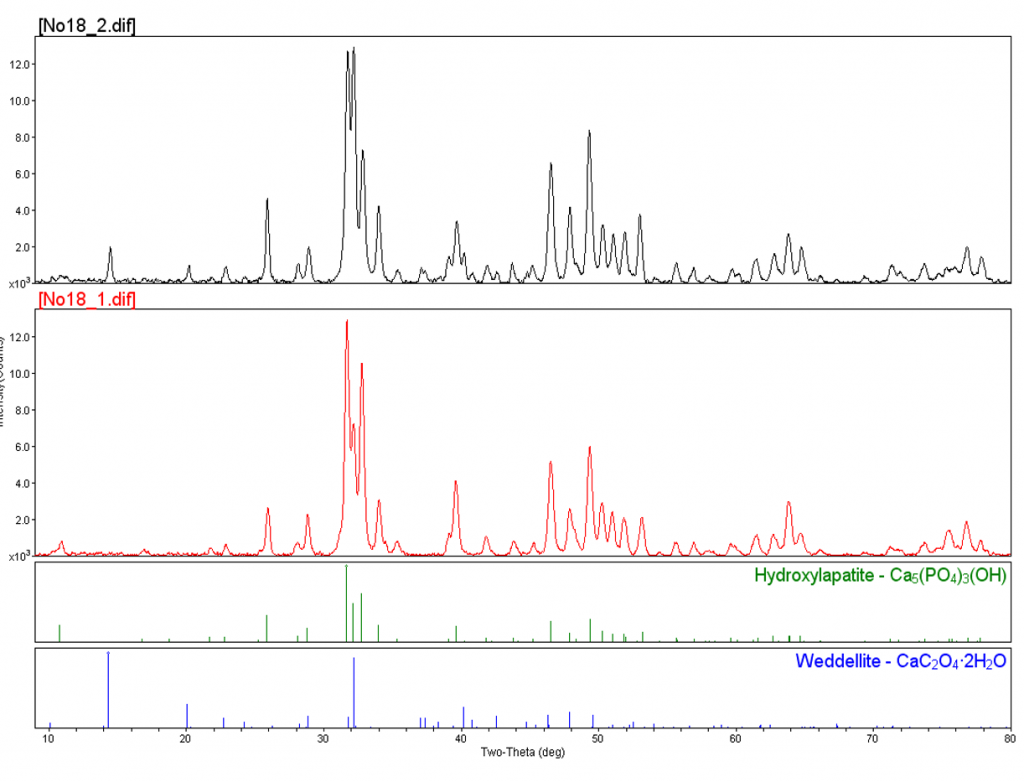
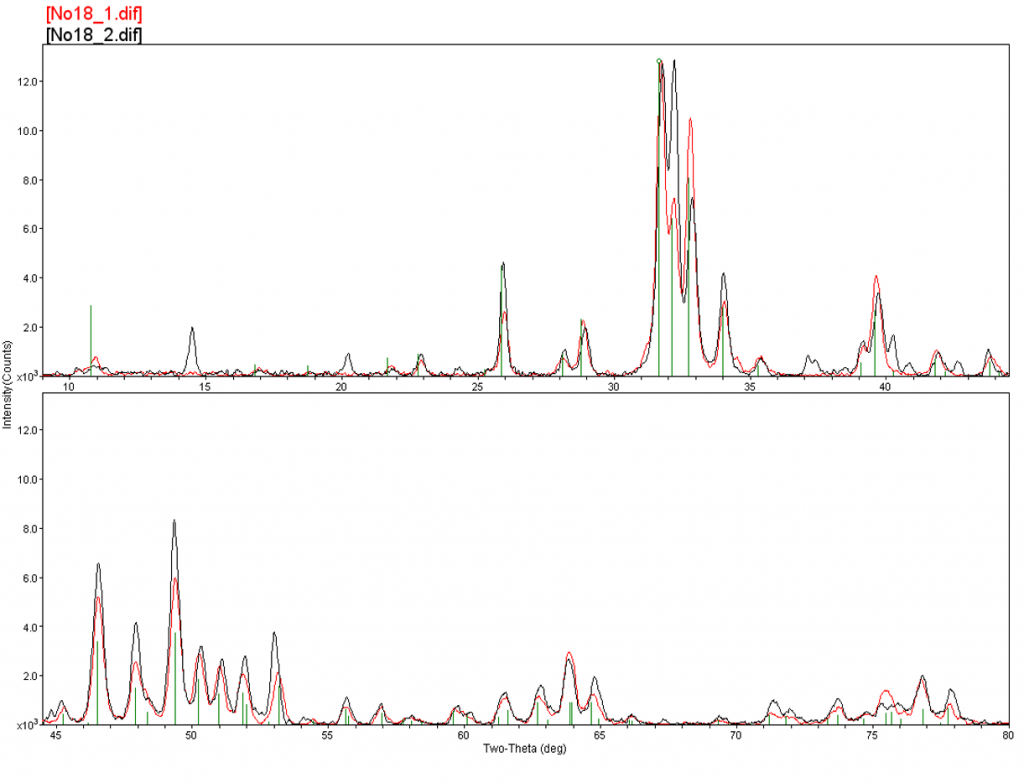
Here’s an example of a large sample. This sample is about a millimeter or two in size. You can see there’s a dark area covering part of the sample. What we attempted to do was to analyze the base material, which was white, and the dark material, to determine if we could distinguish the phases between those two areas. You can see these are overlay patterns, Number 1 being the white material, and Number 2 being the dark material. If we blow this up, you can see the dark material, Number 2 (or the black pattern), there are a few extra peaks overlaid with the Number 1 pattern, since it’s underneath the black material. If we match these to a reference, we see that the underlying material is hydroxylapatite and the black material is a calcium oxalate hydrate. This particle is actually from the presenter’s tooth that fell off during lunch one day. The black material, or the calcium oxalate hydrate, is indicative of tooth decay.
(Click on images to enlarge.)

For the final portion of this seminar, I’d like to talk about particles removed from cross-sections and layered samples. We receive many of these at McCrone Associates. We do several different techniques on particles removed from cross-sections, including optical properties, organic classification, SEM/EDS, quantification by organic methods, and also phase identification.
To give you a quick overview, we will have a cross-section sample, such as this cross-section of paint. We can look at it using SEM/EDS in secondary, and in backscatter, mode, which gives us more of an indication of which elements are present, and number these layers. Once this is determined, we can remove particles for other analyses, such as X-ray diffraction, or others, such as FTIR.
To give an example, here is a test painting where simple store-bought canvas was primed with pigment in two or three layers, then cross-sections were taken from this painting and analyzed by SEM/EDS and light microscopy, and particles were removed for X-ray diffraction. From one area, you can see there are three layers here. The first white layer, we used what was marketed as titanium white; the second yellow area as cadmium yellow light; and the top layer is Prussian blue.
(Click on images to enlarge.)



Samples of each of these were removed in the cleanroom and mounted on pins for XRD. The titanium white showed zincite, barite, and rutile. Rutile is as a form of titanium oxide, and the barite and zincite are also a white pigment, but barite can also be used as a filler; not surprising. The cadmium yellow light was analyzed and showed to be a cadmium zinc sulfide, which is the cadmium yellow pigment. The Prussian blue was analyzed, and this sample did exactly match the ICDD reference for Prussian blue.
(Click on images to enlarge.)
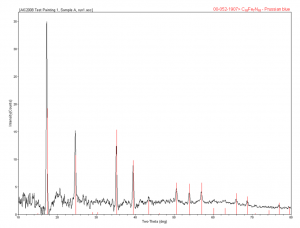
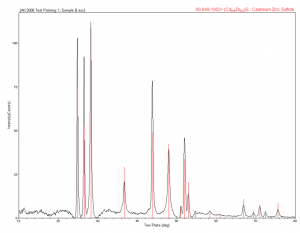
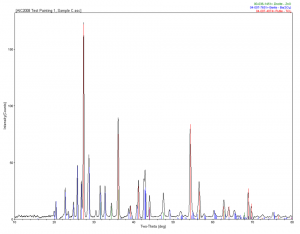
Painting Area 3 also had a few different layers: burnt sienna was mixed with oil, Prussian blue hue watercolor was also applied, as was a top layer of indigo and linseed oil. The burnt sienna in oil showed hematite, quartz and barite, very typical of this type of pigment. The Prussian blue hue showed copper and iron phthalocyanines, which are very typical of Prussian blue pigments. The indigo and linseed oil did only show barite, and indigo is a crystalline material, but perhaps there wasn’t enough in it to be detected by X-ray diffraction, which is usually around a percent or two.

Another layer type of sample is architectural lead paint identification. We receive samples from inside or outside architectural structures in layers to determine if the pigment—the toxic pigment white lead carbonate—is present. We receive particles as chips, dusts, dust wipes, or samples to confirm the presence of this pigment. The polished mounts for these are actually much more layered, and as you can see, some layers are actually white. We will analyze these using SEM/EDS first, to determine which layers are, in fact, lead, and remove particles from those to determine if white lead carbonate is present by X-ray diffraction.
My final example is a 15th century panel painting we did not have here. Here, we removed this one sample from the painting. This sample actually had several layers on it. It was actually a good candidate to get a lot of information from a very small amount of material. This image is the backscatter electron image, and you can see the difference in material from the backscattered electron image when compared to the optical image. To give a few examples, Sample 1: Layer 2 was blue. You can see the blue, and the top of that layer. If you look at the SEM/EDS picture, you can see there’s definitely a darker, I mean more intense, material. EDS showed that this pigment was rich copper, and, in fact, X-ray diffraction showed that it was azurite, which is very typical for this time period.
(Click on images to enlarge.)


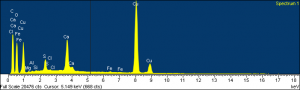
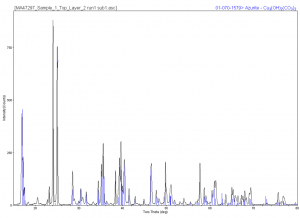
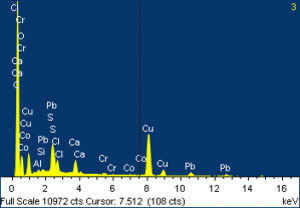
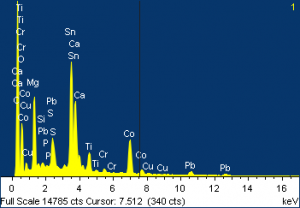
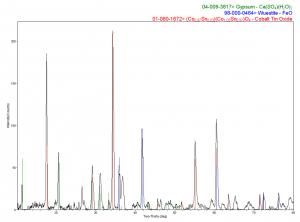
A particle from Layer 4 was analyzed and showed very different elemental compositions: many with lead, some with chromium, and some, in particular, with cobalt and tin. X-ray diffraction of one of these particles showed gypsum, which is most likely from the ground, and iron oxide, and cobalt tin oxide.
(Click on images to enlarge.)
How much material do you need to run an XRD analysis? As you can imagine, you need very little material—a maximum of maybe hundred milligrams, simply for homogeneity. But if it was less than that, even a few milligrams, we could run that as well.
I want thank you for joining us today. I hope this was very informative for you. Please let me know if you have any questions.
CZ: Okay, we have a few more questions rolling in here. Do you want to take a look at those, Joe?
JS: Okay, the first question is: “What is a typical diffraction acquisition time?” This depends on the size of the sample. Typically for pharmaceuticals, for powders like the sample I just mentioned, we typically run them for 15 minutes in duplicate for homogeneity purposes. If we had small particles, say, 10 µm to 20 µm, we may have to run those for half an hour to an hour, depending on how crystalline they are.
CZ: Another question is, “I use dispersive micro-Raman for polymorphs. Does X-ray have advantages over Raman?”
JS: We can also do polymorphs. We get this question a lot for pharmaceuticals, but we would need to have the reference of the polymorphs to determine those. We determine crystal structures from unknowns; from an unknown crystal structure, I guess I should say, but to determine different polymorphs, for instance of an API, we would need to have those references. We can actually have a tabular reference if it’s not the actual sample, and match the sample patterns to the list of peaks, but it is also much better to actually run the standard along with it and compare that.
JS: “Have I used XRD to measure the relative crystalline of the paper?” I see it is from Cindy, who I know—hi, Cindy. Actually, I have not. That would be interesting to see—how crystalline paper is. That is something we could probably do on a small scale. We would probably do a sample size of, maybe, square couple hundred microns would be suitable for us. But, no. We have never measured that, but I don’t see why we couldn’t do that. The only issue might be if the paper is very thick, we would not be able to do it by transmission, because we get a lot of absorption from the paper. Thanks, Cindy.
CZ: We have a couple more questions that just came in. “I use multi-curve resolution for mixtures and polymorphs. Is this possible with X-ray?”
JS: I’m not familiar with the multi-curve resolution. I am not sure if that’s XRD or not. I’m not sure if that is possible with X-ray.
CZ: “Is it possible to run XRD on ophthalmic ointment that has two actives?”
JS: We actually have run XRD samples on ointments. The issue can come up if the excipient, or the gel like ointment, absorbs the X-rays a lot; then we don’t see the API. This also happened with a lot gels we have run before, like topical gels that have APIs in them. But more times than not, we will see the API if it is in a significant amount, which is usually more than a few percent. We can run solutions and ointments with APIs in them with pretty good success.
CZ: Another question: “Sometimes the XRD patterns of single crystal does not match with the simulated patterns from single crystals. What does it reflect? Is it polymorphs?
JS: Possibly polymorphs. This is something when you’re talking about single crystal patterns. If you’re talking about determining crystal structures, again, the indexing is something we don’t do at McCrone. We concentrate mostly on identifying powder samples instead of single crystals, but if the simulated pattern doesn’t match, it would depend on the material, so I’m not sure how to address that.
CZ: It looks like that might do it for the questions. Thanks again for attending today’s webinar. Please visit McCrone.com for a schedule of our future webinars.
Comments
add comment